
The main obstacle in protein engineering is the lack of diversity in training data, which makes it difficult to create high-performance enzyme variations with the desired characteristics and emphasizes the need for better techniques. Here, scientists present a cutting-edge method known as neurofold that overcomes these drawbacks and greatly increases the yield of functioning enzyme variations. NeuroFold uses a multimodal strategy to improve enzyme engineering’s discrimination stages. This method provides a well-informed and diverse sample of the sequence space by utilizing homology, structural, and sequence data. The Spearman rank correlation has increased 40 times as a result of this method, making it possible to quickly produce high-quality enzyme variants like the β-lactamase variants used in this investigation. This pipeline represents an encouraging development in the field of enzyme engineering.
Introduction
The goal of the various previous studies was to design customized enzyme variations with desired chemical characteristics, like better solubility, wider pH stability, increased or lowered reaction rate, and higher thermostability. However, because of the size of the protein sequence space, producing such desired variants is still a difficult operation. Because of their expensive and highly parallelizable assays, conventional experimental techniques like deep mutational scans (DMS) are not acceptable for producing better enzyme variations. A particular study examined the fitness landscape of point mutations in three orthologs of indole-3-glycerol phosphate synthase (IGPS), highlighting the significance of both structural and sequencing characteristics. Vitamin K epoxide reductase (VKOR) missense variations were examined for differences in abundance and activity using multiplex assays in a distinct investigation. These studies highlight the limitations of producing a substantial number of high-quality enzymes due to time-/labor-intensive assays and sub-universal protein sequence coverage.
The Significance of In Silico Protein Design
Latest in silico methods for creating enzyme variations have been developed recently as a result of the rise of deep learning-based models. Different protein language models (pLMs) have shown variable degrees of efficacy in predicting the fitness of enzymes, including ESM-MSA, ESM-1v, and CARP-640m. These more recent methods streamline the experimental bottleneck needed in the past and allow for the faster generation of superior enzyme candidates.
Problems of Deep Learning Models
AlphaFold2 and other structural models’ uneven precision makes it impossible to anticipate changes that affect protein structure. ESM-1v and CARP-640m are two structural models that have demonstrated some degree of accuracy in their ability to distinguish between mutations and orphaned sequences but not much else. Single-sequence models are said to be limited since they are unable to represent co-evolutionary interactions that aren’t implicitly reflected in their model weights. The explicit multiple sequence alignments of evolutionary approaches, such as ESM-MSA, and the implicit capture of information about the protein landscape in their model weights allow them to perform far better when it comes to mutation discrimination. All things considered, the precision with which protein structures can be predicted using deep learning and structural models is limited.
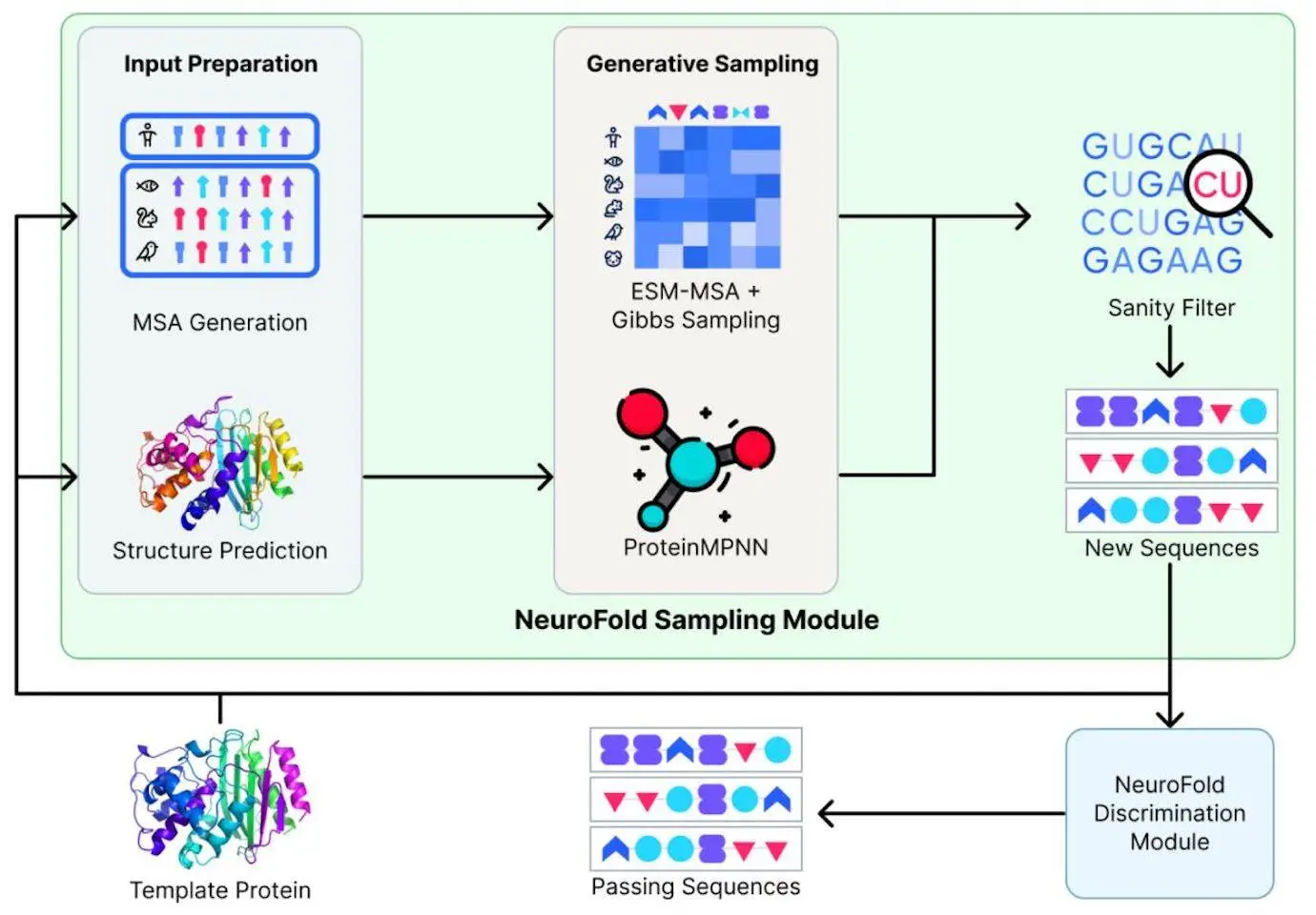
Understanding NeuroFold
NeuroFold is a novel hybrid model that combines evolutionary and sequencing data into a single multimodal strategy. By combining these several data sources, this method goes beyond depending just on one modality, like evolutionary or sequencing data. With the use of NeuroFold, one may more effectively reason about the protein space and provide theoretical constraints that direct the production of workable enzymes. β-lactamase mutants with varied degrees of activity in comparison to the wild-type and improved thermostability have been successfully designed using this method. With its increased coverage of the sequence space, NeuroFold shows promise in generating variations of the enzyme with better stability, more favorable physicochemical characteristics, and faster reaction rates.
Feature Generation Module
NeuroFold makes significant use of the Feature Generation Module (FGM). From an input sequence, the FGM creates an output representation that is utilized by the network as a whole. The two primary parts of the FGM produce properties called MSA and projected structure. The Ensembling Module, which is a collection of models for every modality, receives these features after that. These models’ output is combined based on modality and sent back as an output representation. Whereas AlphaFold2-ptm predicts two protein structures for every input sequence, the MMseqs2 algorithm produces local MSAs. The model’s ability to predict outcomes is enhanced by this improved representation, which biases it to generate more realistic proteins.
β-Lactamase Variant Generation Employing NeuroFold
The purpose of the study was to evaluate how well NeuroFold generated variations of β-lactamase, an important and well-characterized enzyme from Escherichia coli that is used in medicine. The conserved residues implicated in substrate binding and catalysis, such as Ser70, Lys73, Ser130, Asn132, Glu166, Asn170, Lys234, Ser235, Gly236, and Ala237, make up the active site of β-lactamase. It has been shown that mutations at these residues decrease activity. Using the Neurosnap platform, ProteinMPNN was utilized to produce 4,096 different β-lactamase enzyme variants. After that, a collection of viable candidates was generated by applying NeuroFold to filter the resultant variations. For testing based on their anticipated topology and sequence alignment, four of these viable candidates were chosen at random. In order to bias ProteinMPNN and increase the likelihood of obtaining functional enzymes, the catalytic sites of three of the variants (known as Preserved Variants, or PV) were fixed during the ProteinMPNN sampling procedure. Interestingly, sequence identities to the wildtype enzyme ranged from 49% to 56% for all variants chosen for experimental validation, and mutations were evenly spaced over the whole protein sequence. The variations’ anticipated secondary and tertiary structures were almost equivalent despite the low sequence identity. The similarity in projected molecular weight, expected pI (5.14 – 5.94), and amino acid composition indicate that the NeuroFold pipeline is efficient in generating variations with low sequence identity while maintaining significant physical properties.
Conclusion
NeuroFold is a multimodal model that combines structural, evolutionary, and sequencing data to create functional variations of enzymes. When compared to the wild-type, it can produce distinct forms of the β-lactamase enzyme, which can increase activity and thermostability by up to 73%. However, when it comes to working with specific enzyme groupings, such as big proteins, multimeric proteins, and orphaned proteins, NeuroFold is limited. Due to VRAM and compute time limitations, it also has trouble producing variations of big enzymes with more than 2,500 amino acids. Furthermore, NeuroFold’s suitability for proteins, including multimeric enzymes, allosteric binding domains, or metal-binding sites, has not been sufficiently investigated. These restrictions imply that in order to increase its application across a wider spectrum of enzymes and molecular configurations, more research and development are required.
Article source: Reference Paper | NeuroFold is available at https://neurosnap.ai/neurofold
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}