
Traditional drug discovery and development methods are quite expensive, cumbersome, and prone to the biases of experts. A revolution in the field of drug development has emerged in the form of aptamers. Aptamers are single-stranded oligonucleotides (DNA/RNA) capable of binding to biomolecules with high affinity as well as specificity. However, the conventional process of aptamer development called Systematic Evolution of Ligands by Exponential Enrichment (SELEX) is quite expensive, tedious, influenced by the library choice, and usually produces unoptimized aptamers. To add efficiency to aptamer development, researchers from Brock University have developed an AI framework called DAPTEV that generates optimized aptamer sequences for aptamer-based drug development. In this study, the researchers have utilized the COVID-19 spike protein as the molecular target, and the results suggest that DAPTEV excels at producing optimized aptamers with great binding affinities and high specificity that can inhibit the entrance of SARS-CoV-2 virus into our cells.
The Story Behind Aptamers
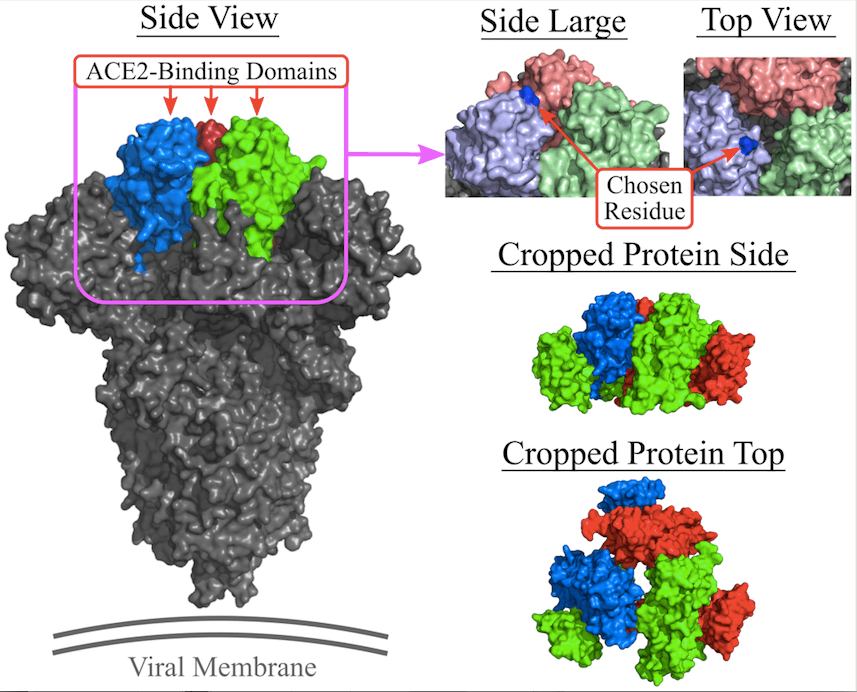
The conditions determining whether an organism is living or non-living become blurry when it comes to viruses. Even though viruses contain genetic material either in the form of DNA or RNA, they are incapable of reproducing by themselves. They have to rely on the host cell’s protein-creation machinery to reproduce viral proteins. Viruses then cause infection by injecting their genetic material into the host cells. Injection of the genetic material is achieved by the binding of viral protein to the cellular receptor with the help of a specialized area on the viral protein called receptor-binding domain (RBD) which has evolved especially for binding to the host cell receptor.
In the case of COVID-19, the viral protein is SARS-CoV-2 spike protein, whose RBD binds the angiotensin-converting enzyme (ACE2) receptor in the lung cell. Conventional drug discovery and development focus on vaccines or therapeutics to combat viral infection. Vaccines offer a preventative approach by preparing the host’s immune system beforehand to attack the virus in case the infection occurs in the future, while therapeutics interfere with the life cycle of the virus once the person has already become infected.
The aptameric approach, on the other hand, binds to the RBD of the viral protein, preventing the viral protein from binding to the host cell receptor, resulting in the virus failing to inject its genetic material. The specialty of aptamers is that they strongly bind to the desired molecular target. Aptamers are synthesized using the SELEX method, which is slow and requires multiple experiments. Also, it has a high chance of providing subpar results as it depends on a random search of top hits, which relies on the original choice of libraries.
Supercharging the Synthesis of Aptamers with AI
These days AI has proven itself to be indispensable in deriving unprecedented insights from data as well as generating new data from input data. AI and machine learning have started getting implemented in small-molecule drug design, however, their utilization in aptamer design has been very limited. This can be attributed to aptamers being an unfamiliar concept in the AI community, the lack of sufficient and reliable data, and the dearth of chemoinformatics tools suited for aptamers.
In their study, researcher Yifeng Li and his team addressed the question of whether AI approaches can propel aptamer design for SARS-CoV-2 with the help of an AI framework called deep aptamer evolutionary model (DAPTEV) that they designed themselves. DAPTEV combines the power of the generating capabilities of deep generative models (DGMs), superior exploration capacity of evolutionary computation, and the quantitative measure of RNA-target binding affinities and utilizes the SARS-CoV-2 spike protein as the target.
The aptamers designed through DAPTEV can serve as the benchmark dataset for the aptamer designing process, and the validation of these aptamers could provide an alternate approach to dealing with the pandemic.
Aptamer Development Methods Before DAPTEV
The old methods for choosing aptamers were confined by intellectual property concerns. With the previous exclusivity no longer in force, fresh techniques emerged. They are:
Aptamer Identification Methods: Traditional computational techniques employed clustering and motif identification to uncover aptamers emerging from SELEX collections. Techniques like AptaCluster and FASTAptamer-Cluster focused on uncovering similarities among aptamer sequences, whereas approaches like MEMRIS and Aptamotif sought to identify recurring structural elements with robust binding properties. Despite their effectiveness in other areas, these approaches struggle to accommodate comprehensive support for secondary structures. An innovative approach known as SMART-Aptamer integrates cluster formation and pattern detection while also addressing the increase in pattern abundance, aptamer groups, and sophisticated three-dimensional architectures.
Supervised Aptamer-Protein Interaction Prediction: Supervised modeling yields reliable predictions of protein-aptamer interactions. With access to a limited quantity of data, standard techniques, and manually designed features were implemented, which resulted in moderate accuracy. These methods involved combining random forest classification and ensemble methods with molecular-based features. Artificial intelligence and RPITER together enable precise prediction of protein-ncRNA interactions. As more data becomes available, similar deep models could be used for aptamer-protein interaction prediction.
Aptamer Generation: Generative models create new DNA and RNA aptamers that bind to specific proteins. Im et al. and Park et al. used generative models with DeepBind to estimate the affinity and specificity of generated sequences. The created sequences exhibited similar structural features to previously discovered ones. Iwano et al.’s RaptGen used a CNN-LSTM encoder and profile hidden Markov model decoder, focusing on variable-length sequences.
Molecular Optimization: The passage mentions DEL (deep evolutionary learning), a multi-objective deep learning framework for small-molecule design. Although it doesn’t work with aptamer data, it inspired the development of DAPTEV, the method being discussed. DEL focuses on molecular optimization and enhancing property distributions. This part of the passage serves as inspiration for the DAPTEV approach.
Comparing DAPTEV to Earlier Methods
- Target Specification:- The approach employed by DAPTEV deviates from prior techniques regarding protein identification. Previous research focused primarily on individual protein molecules, whereas DAPTEV allows for the explicit identification of a target RBD. The researchers have the flexibility to pick any area of a protein and assess its interaction affinity using this new methodology. A more flexible and adjustable methodology is utilized in projecting aptamer-protein relationships.
- Binding Affinity Calculation:- Introducing a fresh perspective on calculating binding affinity, DAPTEV utilizes thermodynamic principles to deliver precise results, unlike previous methods, which relied on estimating binding affinity using approximations by machine learning models such as DeepBind. Employing thermodynamic methods offers a clearer and more precise approach to assessing the intensity of aptamer-protein interactions, which may lead to more dependable forecasts.
- Variable-Length Sequences:- DAPTEV accommodates varied sequence lengths in an innovative manner. Earlier methods centered around creating fixed-length sequence patterns, whereas DAPTEV enables the creation of diverse, adaptable sequence inputs and outputs. Due to this, the generated aptamer sequences can differ in length, offering greater adaptability and authenticity when representing aptamer diversity, which is critical for connecting with multiple protein molecules.
The Mechanism of DAPTEV
The “Deep Aptamer Evolutionary Modeling” (DAPTEV) framework is an innovative approach that blends the power of deep generative models, computational intelligence, and bioinformatics tools. This cutting-edge framework focuses on predicting RNA secondary and tertiary structures as well as RNA-protein folding and docking using Rosetta. With its ultimate goal being enhancing the binding affinity between aptamer sequences and specific protein targets, let’s delve into how this powerful mechanism works:
Dataset Collection and Preprocessing:- The first step of the process involves collecting a dataset that comprises aptamer sequences alongside their corresponding secondary structures. Additionally, docking scores to a target protein are also included in this dataset. In cases where secondary structures are unknown, specialized tools like Arnie can be utilized to compute them accurately. These secondary structures play an integral role in conducting precise docking simulations.
Variational Autoencoder (VAE) Pretraining:- The process of Variational Autoencoder (VAE) Pretraining involves training a VAE by utilizing the given dataset. The VAE consists of two vital processes: encoding and decoding. During the encoding process, aptamer sequences are transformed into continuous embedding vectors, while the decoding process reconstructs sequences from these vectors. The key feature of the VAE is its continuous latent space, which allows for various optimization techniques to be employed.
Evolutionary Operations for Aptamer Optimization:- In this step, an iterative, evolutionary process forms the foundation of the framework. Initially, a population of aptamers is selected from the dataset and encoded into latent vectors. This population undergoes several operations, such as tournament selection, crossover, and mutation, in order to generate new aptamer sequences based on these latent vectors. To evaluate and optimize their performance, the docking score (binding affinity) is used as a fitness metric.
Decoding and Fitness Evaluation:- Decoding the modified latent vectors yields fresh aptamer sequences. The binding potential of these sequences is assessed using computational modeling. The current batch of aptamers is combined with their earlier counterparts, and the most outstanding ones are selected for progression.
Iterative Process and VAE Fine-Tuning:- The iterative process involves continually honing the VAE model by incorporating the recently obtained group. Another cycle of evolutionary manipulation is carried out on the evolved population of aptamers.
RNA-Protein Docking Using Rosetta:- The RNA-protein interaction is modeled by simultaneously folding the RNA and positioning it on the protein surface through Rosetta’s RNP-denovo. Two scoring functions are used: a native Rosetta docking score and a constraint scoring function that guides where the RNA docks on the protein. These simulations provide docking scores for fitness evaluation.
Computing Time Improvement:- To overcome the computational demands of docking simulations, multiprocessing is implemented, allowing multiple simulations to run in parallel on multiple CPUs. This significantly reduces the overall computation time required for the simulations.
An Overview of DAPTEV’s Performance
- In the experimental setup, the docking target for RNA aptamers was the SARS-CoV-2 spike glycoprotein’s receptor-binding domain (RBD). A dataset consisting of known aptamers and a collection of randomly generated sequences were utilized for experimenting. The main objective was to optimize the binding affinity and specificity of the aptamers.
- For comparison, four models were used. They were DAPTEV (with real aptamer data), DAPTEV-X (without real aptamer data), Genetic Algorithm (GA), and Hill Climber (HC).
- Docking Score Optimization: While GA had good docking results, this was largely because it tends to form improperly folded RNA structures. This artificially improved docking scores but compromised structural complexity.
- Balancing Complexity: The research aimed to improve docking metrics and preserve optimal structural features simultaneously. DAPTEV successfully achieved this balance, outperforming GA in terms of producing folded structures without sacrificing docking scores.
- Learning Structural Motifs: DAPTEV’s enhanced folding prowess relative to GA in recent generations suggests that it has effectively learned to recognize and reproduce advanced structural patterns during training, supporting the study’s focal points.
- Querying for New Sequences: DAPTEV successfully generated sequences with outstanding performance in docking simulations. While GA requires lengthy retraining procedures, DAPTEV’s trained model streamlines this process for new aptamer sequences.
- Meeting Set Threshold: All docking scores produced by DAPTEV in the last generation were below the set threshold of 3,500. This indicated that DAPTEV learned and maintained the necessary structural features for effective docking to the target’s RBD.
- Comprehensive Solution: DAPTEV demonstrated impressive performance across the three main areas: scoring optimization, preservation of useful structural elements, and the generation of tailored aptamer sequences through an efficient process.
- Model Comparison: While GA concentrated mainly on optimizing scores regardless of motif conservation, DAPTEV astutely combined these aspects, producing a more inclusive and efficient response.
- Hill Climber Limitation: The hill climber technique often falls short due to its tendency to settle for a low-quality solution too swiftly.
Limitations of DAPTEV
- Limited Starting Data: DAPTEV’s performance was constrained by the availability of initial aptamer data. Only 344 out of 849 known aptamers were utilized due to parameter and computation time limitations.
- Focus on RNAs: The research primarily concentrated on RNAs, neglecting DNAs. A future goal is to incorporate DNA aptamers, necessitating the development of a DNA-protein docking module.
- Secondary Structure Control: While the model requires generated sequences to have at least one connection in the secondary structure, providing more control over the secondary structure’s complexity and connectivity would be advantageous.
- Prediction Accuracy: DAPTEV’s performance can be influenced by limitations in the accuracy of predicting both secondary and tertiary RNA structures.
- Gaussian Latent Space: The assumption of a Gaussian distribution in the latent space might not fully capture the complexity of the problem. Exploring alternative distributions could provide more insights.
- MD Simulation Simplification: While molecular dynamics (MD) simulations offer valuable insights, their reliance on simplified molecular environment models suggests that experimental validation with synthesized aptamers interacting with the target protein is ideal.
Conclusion
Aptamers provide a novel way of combating viral infections. They block the entry of a viral particle altogether, preventing it from entering the cells and proliferating in the body. Adding more to their abilities is their high specificity and binding affinity. Through scientific methods, optimized aptamers can be synthesized conveniently, and with the advent of tools like DAPTEV, their synthesis is further hastened with great improvement in quality as well. Even though DAPTEV has its own share of limitations, considering that it is at an early stage, adjustments and modifications can be implemented to counter the drawbacks. Currently, DAPTEV appears as the most promising aptamer design tool, and there is even scope for future improvements that can certainly catapult the progress in aptamer development, providing humanity with highly effective drugs that can go beyond COVID-19.
Article Source: Reference Paper | DAPTEV code is accessible at GitHub
Learn More:




Neegar is a consulting scientific content writing intern at CBIRT. She's a final-year student pursuing a B.Tech in Biotechnology at Odisha University of Technology and Research. Neegar's enthusiasm is sparked by the dynamic and interdisciplinary aspects of bioinformatics. She possesses a remarkable ability to elucidate intricate concepts using accessible language. Consequently, she aspires to amalgamate her proficiency in bioinformatics with her passion for writing, aiming to convey pioneering breakthroughs and innovations in the field of bioinformatics in a comprehensible manner to a wide audience.











{kind=link}