
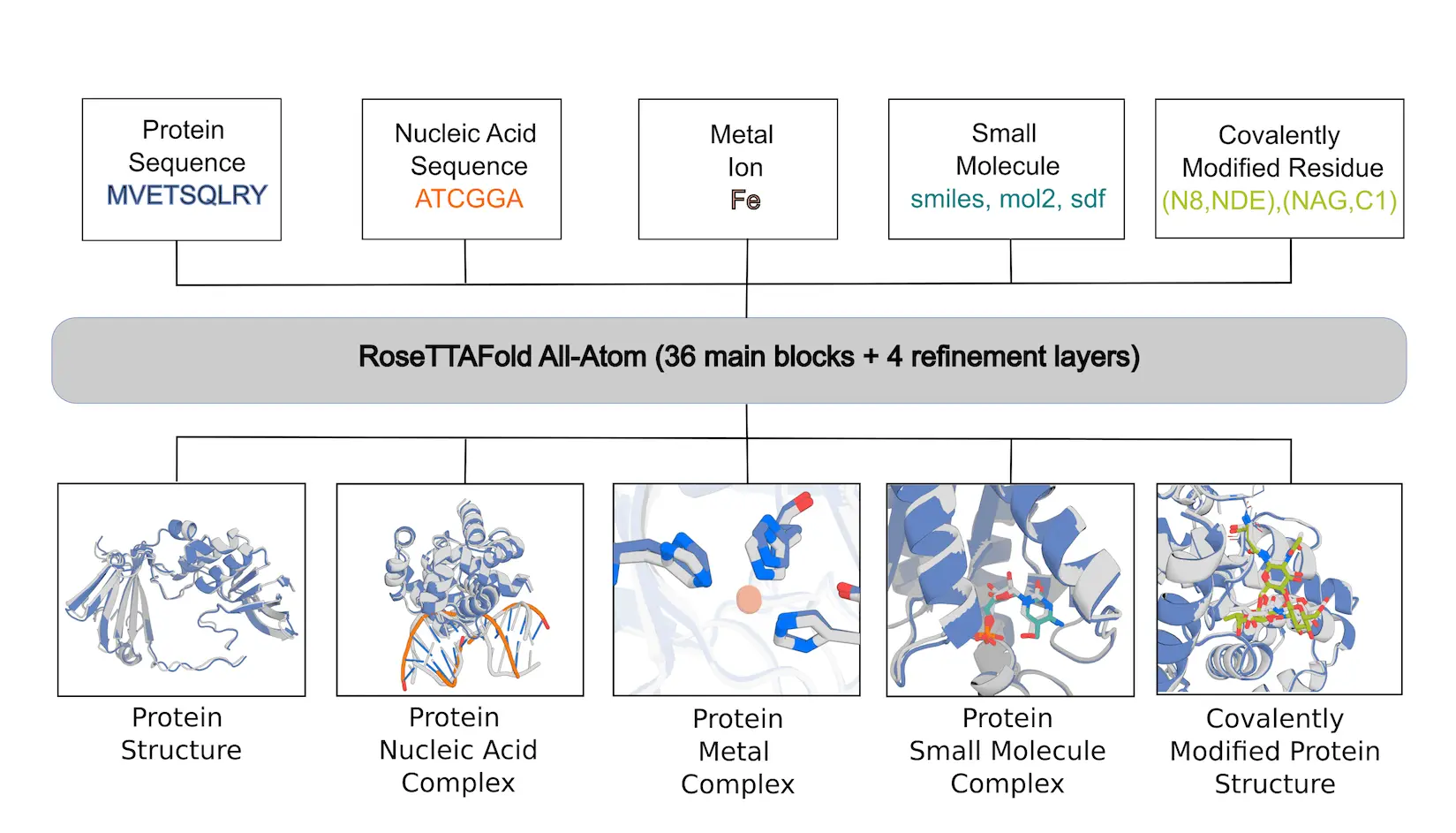
Models like AlphaFold2 (AF2) and RoseTTAFold, a modified structural biology tool, have not been able to replicate well the interactions or covalent changes with tiny molecules that are essential to biological activity. Here, researchers from the University of Washington describe RoseTTAFold All-Atom (RFAA), a deep network that, given the sequences of the polymers and the atomic bonded geometry of the small molecules and covalent modifications, is capable of modeling complete biological assemblies containing proteins, nucleic acids, small molecules, metals, and covalent modifications. RFAA is a protein structure prediction approach with good performance that was trained using the Protein Data Bank (PDB). Comparable to AF2 and CAMEO, RFAA can dock tiny molecules with a flexible backbone with similar accuracy. It also predicts proteins with covalent modifications and assemblages of numerous nucleic acid chains and tiny molecules with a reasonable degree of accuracy. By fine-tuning diffusive denoising jobs and creating binding pockets by constructing protein structures around tiny molecules and non-protein molecules, RFdiffusion All-Atom (RFdiffusionAA) is developed. It is anticipated that these techniques will be helpful for creating and modeling intricate biomolecular systems.
Introduction
Deep neural networks like AlphaFold2 and RoseTTAFold are highly accurate at predicting protein structures. However, during transcription and translation, proteins frequently interact with other proteins, tiny molecules during metabolism, and DNA and RNA. It is still difficult to model biomolecular assemblages made up of random tiny molecules, covalently modified amino acids, polypeptide chains, and nucleic acid chains. One strategy is to use AF2 or RF to model protein chains and then use classical docking techniques to incorporate non-protein components. Although RoseTTAFold nucleic acid (RFNA) has been developed to mimic nucleic acids as well as proteins, the variety of potential tiny molecule components makes modeling biomolecular systems in general difficult. In addition to having a significant impact on structural biology and drug discovery, an approach that can reliably predict the three-dimensional structures of biomolecular assemblies from knowledge of constituent molecules could pave the way for deep learning-based protein-small molecule assembly design.
Insights into challenges faced in the initial developmental phase of RFAA
In generalized biomolecular system modeling, a more significant difficulty is the development of a structure prediction approach for a biological unit, such as proteins, nucleic acids, tiny molecules, metals, and chemical changes. Current protein structure prediction networks can be expanded to represent nucleic acids and view proteins as linear chains of amino acids. It is unclear, though, how to model many tiny molecules as a linear sequence because they are not polymers. An atomic graph model of small molecules and protein covalent changes was coupled with a sequence-based description of biopolymers (proteins and nucleic acids) in order to get around this restriction. With this method, the computational challenges of modeling whole proteins at the atomic level should be overcome.
Understanding RFAA
RoseTTAFold All-Atom is a neural network that can accurately model a wide range of biomolecular assemblies containing a diverse range of non-protein components. It can make high-accuracy predictions on protein-small molecule complexes, with 32% of CAMEO targets predicted under 2Å RMSD. RFAA also predicts 46% of recently solved covalent modifications to proteins under 2.5Å RMSD. This method surpasses previous methods in generating accurate models for complexes of proteins with two or more non-protein molecules. RFAA achieves similar protein structure prediction accuracy as AF2 and protein-nucleic acid complex accuracy as RFNA.
Understanding RFAA architecture
The network architecture, which is based on the RoseTTAFold2 (RF2) protein structure prediction network, improves predicted structures through hidden layers by utilizing 1D sequence information, 2D pairwise distance information from homologous templates, and 3D coordinate information. The network displays arbitrary tiny molecules as atom-bond graphs and preserves representations of protein and nucleic acid chains. Each non-polymer atom’s chemical element type is fed into the 1D track, and the chemical bonds between atoms are encoded in the 2D track. Information about planarity, angles, bond lengths, and chirality can be found in the 3D track. The sign of the angles between the atoms around each chiral center is specified by the network in order to encode stereochemistry information in the third track. The gradient of the departure from ideal values is calculated and supplied to the next block as an input feature.
The network predicts the amino acid and base sequence order using molecular graphs, which are permutation invariant. For bases and amino acids, the network uses relative position encoding; for atoms, it does not. Along the 3D track, the network coordinates and generates rotational and translational updates to each frame orientation. The 3D track is extended with coordinates for heavy atoms that move independently based on anticipated translational adjustments. The entire system is shown as a disjointed gas of nucleic acid bases, free-moving atoms, and amino acid residues that are converted into physically believable assembly structures by the network. For parameter optimization, an all-atom variant of the Frame Aligned Point Error (FAPE) loss is created. In order to help users discover high-quality predictions, the network additionally predicts pairwise confidence (PAE) and atom and residue-wise confidence (pLDDT).
Predicting Protein-Small Molecule Complexes with RFAA
Two difficult modeling problems have led to the development of RFAA, a deep learning approach for protein-ligand docking: generic protein-ligand docking problems and simultaneously docking many small molecules or nucleic acid chains. The RFAA server was added to the blind CAMEO ligand docking evaluation, which makes predictions on all structures that are added to the PDB every week utilizing a number of servers. To determine which predictions were true, the network’s anticipated pairwise error (PAE Interaction) between protein chains and small molecule chains was employed. When modeling protein-small molecules in CAMEO, RFAA consistently performed better than other servers; 32% of situations (< 2Å ligand RMSD) were effectively modeled, compared to 8% for the Vina server. RFAA is capable of modeling higher-order biomolecular complexes involving several proteins, small molecules, metal ions, and nucleic acids because it can simultaneously predict interactions between proteins and multiple non-protein ligands in a single forward pass.
In protein-ligand docking approaches, the ability of a method to dock a tiny molecule with a protein target is determined by its crystal structure. A deep learning-based docking tool called RFAA performs better than DiffDock, correctly predicting 42% of complexes as opposed to DiffDock’s 38%. When the bound protein structure and pocket residues are known, physics-based techniques like AutoDock Vina perform better than RFAA. The increased prediction accuracy of RFAA for protein structures is a result of its ligand context training. Although it does not generalize to new clusters, the network performs better for clusters that overlap with the training set. Additional training on larger datasets could be required to produce consistently good predictions for novel protein-small molecule complexes. For complexes with high projected affinity, a stronger association was observed between prediction accuracy and physically based correlates of protein-small molecule interaction affinity.
RFAA with De Novo Small Molecule Binder Design
One of the biggest challenges in protein design is creating proteins that can bind tiny molecules. In the past, molecules have been docked into a huge collection of native or carefully selected protein scaffold structures. Diffusion techniques can produce proteins that bind with significant affinity and specificity when applied to a protein target, as demonstrated by recent work. Because existing deep learning methods cannot simulate the interactions between proteins and ligands, they are not suitable for small molecule binder creation. Although no experiments have been conducted to validate RFdiffusion, a heuristic technique, it promotes pocket formation with shape complementarity to target molecules. To optimize advantageous interactions, a broad strategy that creates protein structures around tiny molecules and non-protein targets might be useful.
A diffusion model called RFdiffusion All-Atom (RFdiffusionAA) was created to facilitate protein design in the setting of non-protein biomolecules. The use of the protein-small molecule dataset trains a denoising diffusion probabilistic model (DDPM) initialized with RFAA structure-prediction weights to denoise distorted protein structures conditioned on biomolecular contexts. The distribution of proteins is taught to the model conditioned on the biomolecular substructure, which includes small molecule conformation. At each denoising phase, the model updates coordinates and orientations, generating proteins by first initializing a Gaussian distribution of residue frames with randomized rotations around a fixed small molecule motif. In order to increase the number of connections that small molecules and binders make, the model also looks into the employment of auxiliary potentials to alter trajectories. When RFdiffusionAA binders are evaluated in silico, the results surpass those obtained with protein-only RFdiffusion utilizing a heuristic attractive/repulsive potential. Diffusion of radiofrequency with respect to AF2 predictions derived from a single sequence, AA’s small molecule binders exhibit a high degree of consistency. Furthermore, the model demonstrates a notable capacity for generalizing not only protein shape and structure but also interactions with non-protein targets.
Conclusion
A neural network called RoseTTAFold All-Atom (RFAA) is capable of accurately modeling a variety of biomolecular assemblies with a wide range of non-protein components. With 46% of recently solved covalent modifications predicted under 2.5Å RMSD, it can produce highly accurate predictions on protein-small molecule complexes and covalent changes to proteins. When it comes to producing precise models for protein complexes containing two or more non-protein components, RFAA outperforms earlier techniques. The network can predict proteins and ligands that are significantly different from those in the training dataset and learns broad principles about protein-small molecule complexes. For the purpose of drug development, RFAA’s accuracy must be significantly improved in the near future.
Article source: Reference Paper | Reference Article | RoseTTAFold All-Atom is available on GitHub
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











 is a deep network capable of modeling complete biological assemblies containing proteins, nucleic acids, small molecules, metals, and covalent modifications.){kind=link}