
It is still very difficult to construct complex protein folds from scratch using only computational methods. École Polytechnique Fédérale de Lausanne (EPFL) scientists create intricate folds and soluble counterparts of essential membrane proteins using a powerful deep-learning pipeline. Researchers show that the structural characteristics of unique membrane topologies, including those from G-protein-coupled receptors, may be reproduced in solution. These topologies are not present in the soluble proteome. Designing complex protein topologies with functionality derived from membrane proteins has yielded high experimental success rates. These designs show excellent thermal stability and extraordinary design precision, which may open up new avenues for drug development. The functional soluble fold space was shown to have the ability to expand when the soluble counterparts were functionalized with native structural motifs.
Introduction
A key step in developing artificial proteins with novel functions is protein design, which broadens the scope of nature’s molecular apparatus. Historically, physics-based methods such as Rosetta have been employed; however, they necessitate a great deal of experimental screening and optimization, which makes the creation of functional proteins with intricate structural topologies challenging.
Lately, protein structure prediction pipelines such as AlphaFold2 (AF2) have reached previously unheard-of levels of accuracy. Exploring the sequence space and finding proteins with stable topologies and new functionalities has become more viable thanks to deep learning-based techniques. These techniques have proved useful for a number of applications, such as the use of diffusion models to create novel designable backbones, oligomeric protein assemblies, and different protein topologies.
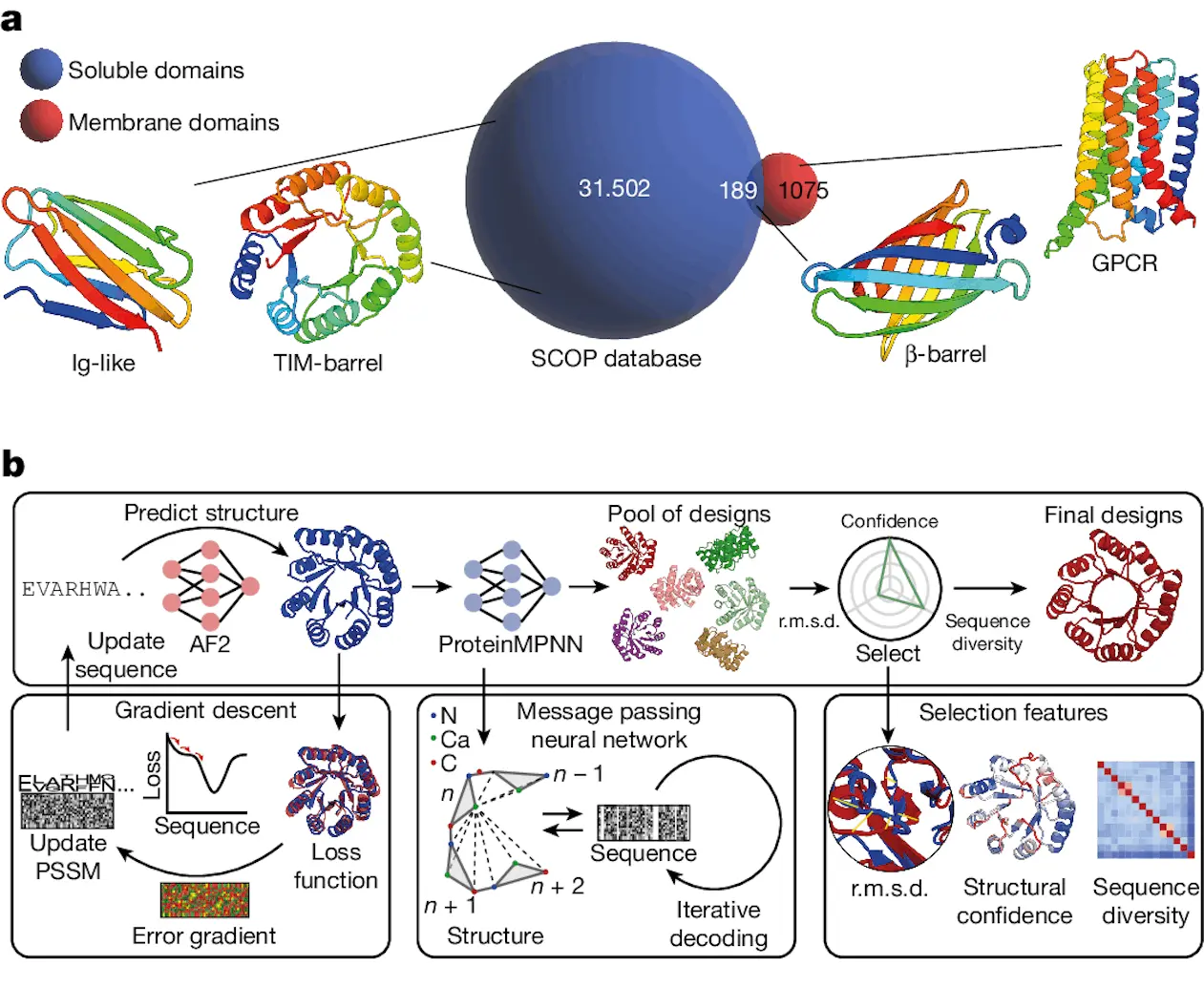
Developing novel protein functionalities requires designing protein folds with intricate structures, such as huge sizes and non-local topologies. An examination of the Structural Classification of Proteins (SCOP) database showed that proteins in the cell membrane environment and the soluble proteome are segregated. Of the 1,075 membrane proteins, only 189 folds were present in both settings and had distinct topologies not observed in soluble form.
This calls into question whether topologies of integral membrane proteins possess basic structural characteristics that prevent them from existing in the soluble fold space. In order to achieve a de facto-fold expansion of the soluble proteome and to open up possibilities for the design of new functionalities employing these hitherto unattainable protein folds, the study looked into engineering membrane folds as soluble equivalents.
Computational Design of Soluble Protein Analogues
Researchers created a novel computational pipeline for reliable de novo protein design in this study. Developing a reliable computational model for intricate protein folds is still a challenging task. Here, researchers introduce a deep learning-based computational method that creates high-quality protein backbones and facilitates the effective search for non-natural sequences for a range of protein topologies.
Because of its flexibility and generalizability, the AF2seq-MPNN-based computational framework eliminates the need for laborious parametric and symmetric design constraints or fold-specific retraining. Researchers achieved excellent experimental success rates in terms of soluble and folded designs by designing and characterizing various folds that have proven extremely difficult to produce with existing techniques.
The structural characterization of computational models showed that protein topologies, both the overall fold and minute details of side-chain conformations, could be designed with a high degree of precision. Extending the soluble fold space and facilitating the design of protein topologies observed in membrane contexts were the goals of the computational approach that was tried. Rhomboid protease and GPCR, two of the three membrane-fold analogs with extremely complex helical topologies, were among the three that were created. The experimental structures demonstrated a high degree of accuracy in the design process, with soluble replicas of the standard seven-helix GPCR fold and the rhomboid protease fold successfully generated. This revealed that many membrane protein folds may be easily constructed in soluble form and often adhere to the same design principles as soluble protein folds.
Computational Design Achieves High-Precision Protein Structures
Membrane protein soluble analogs open up a large range of folds not found in the soluble fold space, which can improve the designability of functional proteins. Analogs of the original membrane proteins that preserve their native characteristics, like ligand-binding or enzymatic activities, speed up research on their roles in soluble forms more readily accessible to biochemical processes. Incorporating native structural patterns has shown to be a promising approach.
The process of developing functional proteins, such as soluble GPCR medication mimics, is known as protein development. It is possible to construct these analogs in particular functional states, which makes it possible to launch small-molecule or antibody discovery efforts. Since the design method is so precise, it is possible to create conformationally specific designs for both active and inactive GPCR states, which are distinguished by minute conformational variations. By using this method, membrane-soluble analogs with native functional characteristics can be produced, which may be essential for advancing the discovery of novel medications and treatments that target difficult protein classes.
The creation of novel drugs and treatments aimed at these difficult protein targets may be greatly aided by the production of membrane-soluble analogs possessing native functional characteristics. Effective exploration of novel sequence spaces, production of viable proteins, and growth of the designable fold space are all made possible by the use of high-quality structure representations in computational protein design through the application of deep learning. This strategy may affect the ability to create functional proteins.
Conclusion
A customizable framework based on AF2seq-MPNN has been designed to build complicated protein folds through a computational technique based on deep learning. This method produces high-quality protein backbones by efficiently searching for non-natural sequences for different protein topologies. The computational model demonstrated good accuracy in overall fold and side-chain conformations, achieving high experimental success rates in soluble and folded designs. The method also made it possible to create protein topologies analogous to those seen in membrane environments, proving that membrane protein folds adhere to the same design guidelines as soluble protein folds.
Article Source: Reference Paper
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}