
Language models have made a new era of biological sequence modeling possible. However, it is still difficult to extract significant sequence-level embeddings from these models. ProteinCLIP, which employs contrastive learning between a protein’s amino acid sequence and curated literature detailing its function, is introduced by Stanford University researchers in this paper. Thus, ProteinCLIP learns to take the sequence embedding of a pre-trained protein language model and modify it to produce a function-centric embedding. ProteinCLIP enables state-of-the-art performance across a range of difficult tasks in protein studies, demonstrating the efficacy of multi-modal learning in biological situations. Predicting protein interactions and correctly identifying homologous proteins in spite of poor sequence similarity are examples of this. This method enhances the usability of large models by isolating important signals.
Introduction
A potent tool for researching biological sequences, such as protein, DNA, and RNA sequences, is large language models, or LLMs. Numerous outcomes have been attained by these models, such as the ability to predict the impact of non-coding mutations on gene expression, the structure of proteins derived from amino acid sequences, the transcriptomic effects of gene knockouts, the identification of homologous proteins, and even the synthesis of new proteins. These models have shown themselves to be an effective resource for comprehending biological sequences.
The training process of biological language models, which includes completing missing nucleotides or amino acids and concealing random locations within a biological sequence, is mostly responsible for the models’ performance. This easy challenge teaches complicated sequence correlations that can be utilized to solve more difficult problems, like predicting protein binding and folding. This method, however, ignores the wealth of information that scientists have amassed over many years regarding proteins and their purposes. In addition to the sequence correlations that biological language models learn, recent research indicates that natural language text can serve as an additional representation of the compositional structure of a biological molecule. By combining these two methods, biological language models can be enhanced.
In machine learning, complementary viewpoints are being utilized more and more to create richer models. Models that allow variable query patterns are trained using textual descriptions and visuals via techniques such as contrastive learning. These models provide the basis for both biology-focused machine-learning projects and generative applications. These techniques, however, are conceptually more limited because they connect particular metrics to direct experimental relationships, which restricts their use in larger machine-learning communities.
Understanding ProteinCLIP
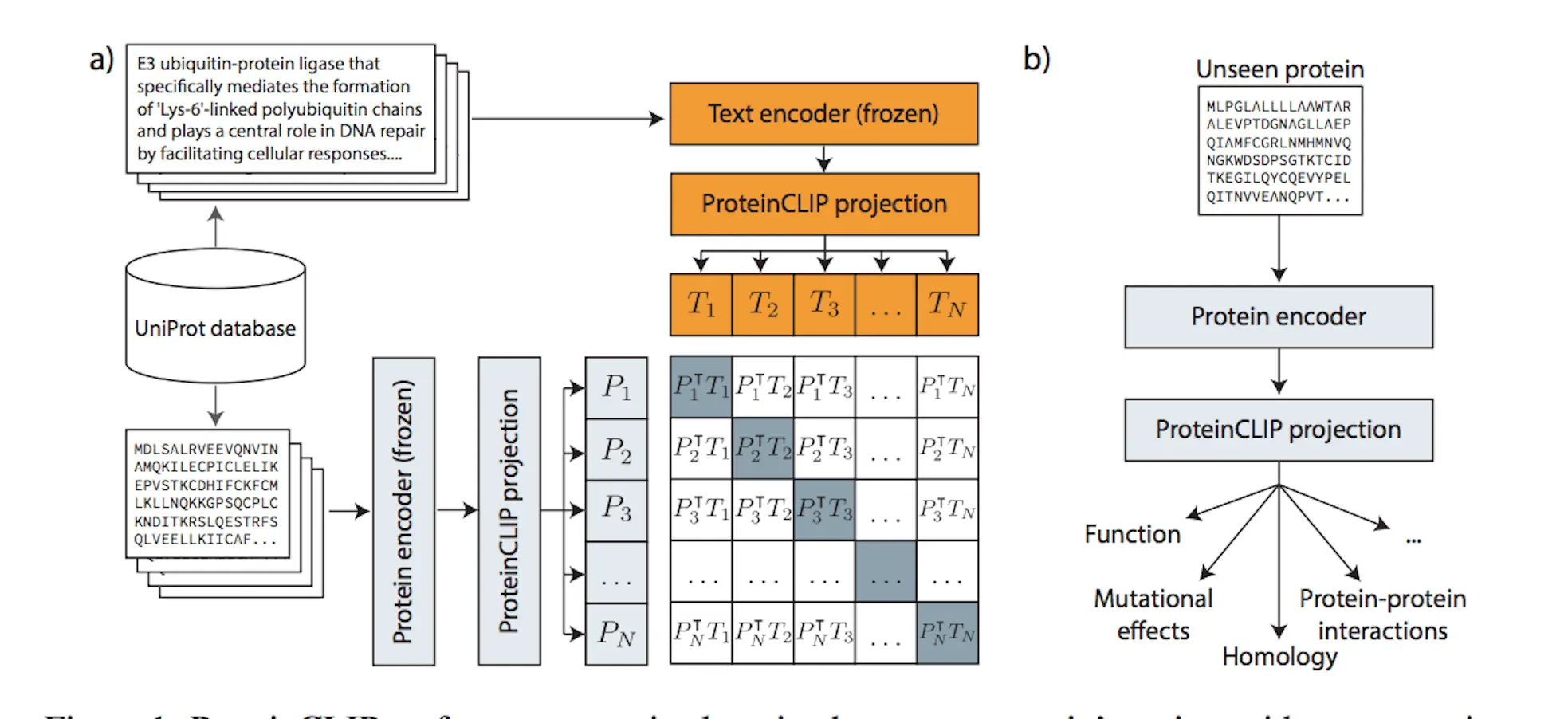
The novel ProteinCLIP method combines natural language models with a particular class of biological language models, namely protein language models. A common embedding representation between protein amino acid sequences and textual function annotations of those proteins is produced by these models, which were trained on amino acid sequences. ProteinCLIP uses contrastive learning to reconcile amino acids with their emergent function, paving the way for subsequent uses such as the creation of sequence-level embeddings that are sensitive to changes in amino acids, the prediction of interactions between proteins, and the identification and annotation of unknown sequences. This versatile method may be used with any protein language model and shows that focusing and extracting significant information from pre-trained biological language models is both effective and computationally efficient when knowledge from many modalities is combined.
Methodology of ProteinCLIP
ProteinCLIP is a contrastive methodology that aims to reconcile textual embeddings of matching protein function descriptions given by a natural language model with amino acid embeddings obtained by a protein language model. It learns to condense a more concise, efficient representation of a function by rearranging the embedding that a protein language model has previously learned rather than fine-tuning the language models it is based on. In order to achieve state-of-the-art findings in predicting proteins’ global response to mutations, identifying protein interaction partners, and locating homologous proteins with minimal sequence similarity, ProteinCLIP is very good at condensing amino acid embeddings towards a more function-centric view. Sequence and function embedding alignment is expected to highlight signals pertinent to homology and identify functional alterations. As shown with SARS-CoV-2 proteins, these advancements in protein annotation pipelines can be used as a foundation for efficiently researching and comprehending biological systems in silico, as well as for large-scale screening of novel protein designs for desirable behaviors.
Limitation of ProteinCLIP
A protein-based machine learning model called ProteinCLIP shows notable improvements in performance across a range of applications, although it is not without limitations. Gains in performance are not possible in tasks unrelated to protein function because of its emphasis on protein function. For example, the prediction of interactions between small compounds and proteins was not improved, probably because structural information was used instead of functional annotation. Furthermore, ProteinCLIP uses average pooling, which has drawbacks prior to applying contrastive learning. Hardware limitations led to the huge network that was produced by alternative methods, such as training an attention-based aggregation mechanism. More research should concentrate on creating contrastive methods with less innocent pooling techniques to achieve higher performance advantages.
Conclusion
ProteinCLIP is a valuable tool for the scientific community since it improves performance on several important protein annotation tasks and provides a more comprehensive understanding of why multi-modal modeling works well in biological contexts. Indeed, ProteinCLIP shows that scientists in the future may extract even more power from these already potent models by carefully combining self-supervised pre-training procedures in a multi-modal approach, despite the fact that these strategies have proven immensely successful for modeling biological and natural text sequences. More importantly, ProteinCLIP shows that we are far from realizing the full potential of these models and that doing so does not always require the enormous computational inputs that come with training ever-larger language models.
Article Source: Reference Paper | ProteinCLIP code is available for the community on GitHub.
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}