
Transposable elements (TEs) supply genomes with both coding and non-coding sequences and have significant roles in evolution. However, the extent of TE annotations in each species has limited the identification of TE-derived functional components to reasonably intact TE sequences. Here, scientists from the École Polytechnique Fédérale de Lausanne, Switzerland, present a novel method that they used to probe several ancestral genomes reconstructed from hundreds of species in order to find degenerate TEs (degTEs) that had not yet been annotated. The study reveals that degTEs are essential for transcription because they support several transcription factor binding sites and cis-regulatory elements, such as the KRAB zinc-finger proteins. A unique methodology for a full-scale investigation of TE co-option events is published, along with unannotated chimeric transcripts between human genes and degTEs, which contributes to our knowledge of these occurrences.
Understanding Transposable Elements
Transposable elements are those genetic units that can change their position within their host genome. After TEs were discovered in the mid-1900s, Barbara McClintock named them “controlling elements” to describe their capacity for regulation. Twenty years later, Eric Davidson and Roy Britten proposed that TEs are essential to the construction of regulatory networks. The idea that TE-derived proteins, non-coding transcripts, and other cis-acting elements are essential to the evolution of genome control and architecture has recently been confirmed by mounting data. In the assembly of the human genome, TEs and repetitive elements, such as DNA transposons, long terminal repeats (LTRs), and interspersed nuclear elements (LINEs and SINEs), account for 54% of genomic DNA. However, due to mutations, the majority of sequences in the human genome are transposition incompetent; only 1:1,000 integrants, such as L1, Alu, SVA, or HERV-K, remain active. Transposition-incompetent TEs are not just parasitic sequences but also co-opt regulatory elements and give rise to new genes. They are spliced into genic transcripts, resulting in TE-gene chimeric transcripts or transpochimeric gene transcripts (TcGTs). Each lineage has distinct TE subfamilies, contributing to lineage-specific genomic innovations, as each lineage has acquired distinct TE subfamilies during evolution.
Introduction
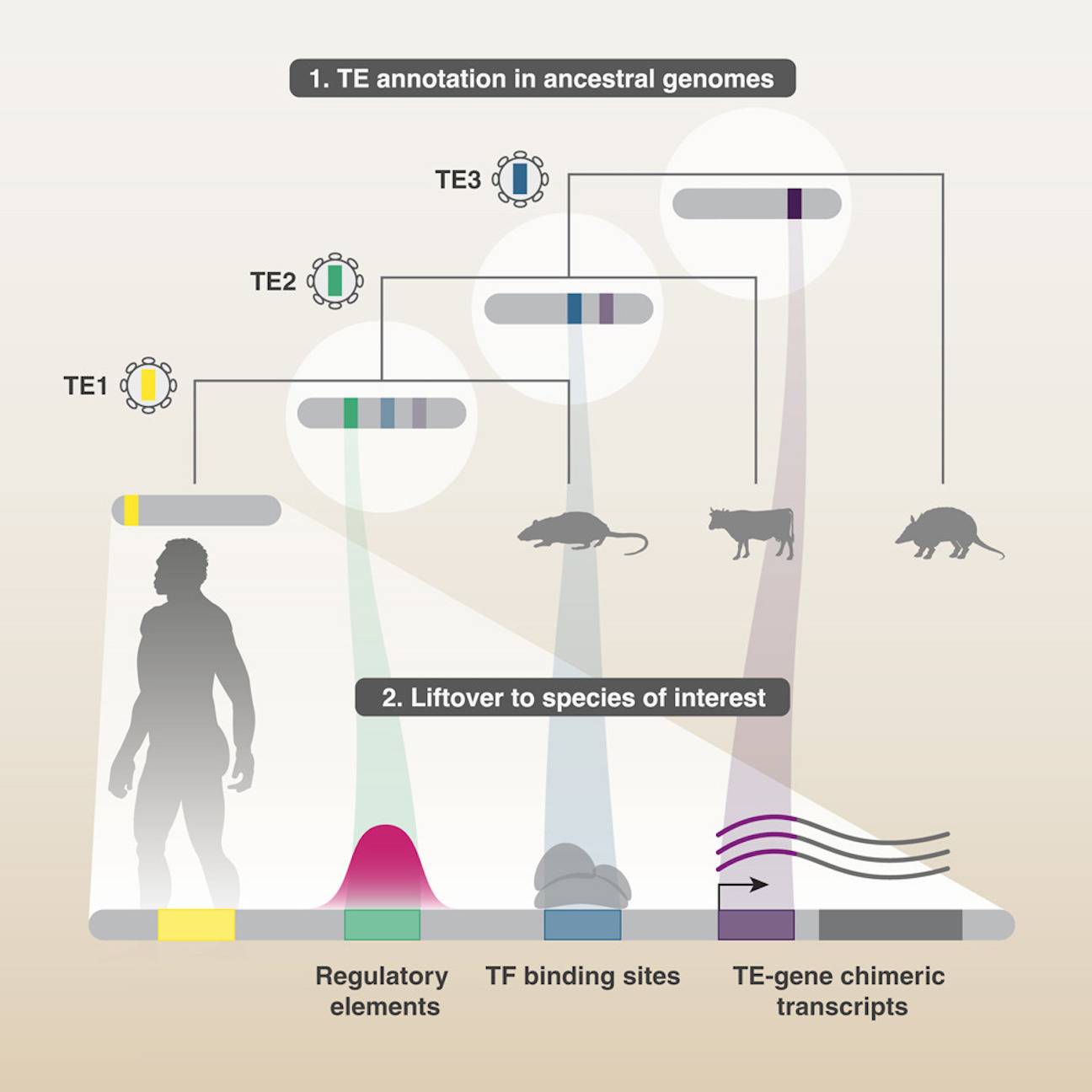
The gold-standard method for annotating TEs in an assembled genome is repository-based annotation, best exemplified by RepeatMasker. This software scans a genome and identifies loci that significantly resemble one of the consensus sequences registered on a TE repository like Repbase or Dfam. However, this method is limited to discovering TEs that retain high sequence similarity to the consensus and might miss highly degenerate TEs, typically evolutionarily old elements. Whole-genome sequencing from a growing number of species—more than 600 vertebrates—has been made available recently thanks to initiatives. Reconstructed Ancestral Genomes (RAGs), as defined by Armstrong et al., are a scalable technique for aligning a large number of genomes by reconstructing their ancestral sequences. For the L1PA6 subfamily, this approach has received some confirmation; however, verification for other TEs has not been conducted.
In this study, researchers discover a unique approach that makes use of numerous RAGs to find previously unannotated TEs; the human genome has been discovered. This technique finds transcription factor binding sites (TFBSs) and cis-regulatory elements (CREs) in chimeric transcripts and increases the overall TE coverage by 10.8%. Additionally, integrants of significant TE classes that were previously unannotated are revealed by this method.
Identifying degTEs
In this paper, researchers present a versatile method using multiple RAGs to identify degTEs. Because the divergence time of TE integrants in older ancestral genomes is shorter, their initial emergence is closer to consensus. However, TE subfamilies that emerged later in evolution are absent from RAGs. Several RAGs ancestral to a species of interest (SOI) are employed to retrieve TEs that originated at widely spread evolutionary time points. TEs are mapped from RAGs and SOI onto the SOI genome to find their corresponding locations. After comparing lifted-over TEs to SOI TEs, integrants that partially overlap are combined to expand the SOI genome’s existing integrants.
Contribution of degTEs to chimeric transcripts
Non-canonical chimeric transcripts (TcGTs) are a result of the contribution of unique sequences by TEs, including transposition-incompetent integrants, to host gene transcripts. TcGTs can target microRNAs, start transcription, and insert non-canonical coding sequences. By creating TE-driven TcGTs, TEs in mammalian embryos specify the expression of host genes at particular stages. Two hundred twenty-two non-canonical exons involving at least one degTE were found in human embryos’ RNA sequencing data, which allowed researchers to investigate if degTEs are involved in TcGTs expressed in human embryos. Up until the 8-cell stage, a large number of TcGTs exhibit significant expression levels and are important isoforms in their host genes. In the developing embryo, TcGTs associated with DegTE manifest themselves differently. This gene’s first annotated exon is 37 kb upstream of a non-canonical exon produced from a MIRb degTE, which is the source of a TcGT that starts AKTIP transcription. This suggests that some degTEs also form TcGTs by either driving their expression or being included as a non-canonical exon.
degTEs contributing to regulatory elements and TFBSs
The purpose of the study was to ascertain if KZFPs, TE-derived CREs, and degTEs contribute to the human genome. Eighty-two thousand four hundred seventy-four cCREs, or 8% of each cCRE group, were found to be related to degTEs by crossing the genome-wide candidate CRE (cCRE) annotation with the upgraded hg38 TE annotation. For every TF, 6,000 TFBSs connected with degTEs were found to assess if degTEs contribute to TFBSs. The largest fraction of degTE-derived TFBSs was discovered in ZMYM2 and its paralog, ZMYM3, which are components of the co-repressor of repressor element-1 silencing transcription (CoREST) complex.
Major TFs that identify TEs are KRAB-domain-containing zinc-finger proteins (KZFPs), which typically bind TE subfamilies that share an evolutionary age. According to the study, evolutionarily old KZFPs are more likely to have degTE-overlapping peaks. KZFPs that were previously found to target evolutionarily ancient TE subfamilies share many binding sites with degTEs that belong to the same subfamilies.
In order to investigate the evolution of KZFP-bound degTEs, the study concentrated on ZKSCAN1, a KZFP that significantly binds elements in the L2 family. The L2c consensus was more closely aligned with the matching RAG sequences than the degTE sequences, according to a multiple sequence alignment. The previously unidentified relationship between cCREs TFBSs and TEs was made clear by the retrieval of degTEs via the examination of RAGs.
RAGs improved the annotation of human TEs by impartially identifying degTEs.
Without significantly altering the distribution of TE classes, the addition of hg38 TEs to the union of degTEs improved the annotation of TEs in the human genome by 10.8%. MamSINE1, MamRTE1, and L2a are examples of recently found integrants; L2d is an extension of an already-annotated insert. Except for a small but notable bias towards areas with fewer TEs already identified in hg38, they were spread out among all chromosomes.
Between the common ancestors of mammals and eutherians, TE subfamilies gave rise to 97% of the degTEs. For these ancient TEs from hg38, the increased annotation produced a 10%–70% increase in coverage. With each subfamily adding about 10 Mb, the L2 and MIR subfamilies that formed during this evolutionary period play a major role in the new annotation.
By using a separate TE annotation tool and applying it to other species, the pipeline’s resilience was evaluated. The three extra species had a 23%–33% increase in coverage thanks to the improved annotation; this gain was not proportionate to the genome sizes or the amount of TE coverage that had previously been annotated. With low FPRs and FDRs, the increased annotation method recovered evolutionarily ancient TEs with higher similarity to the consensus in the RAGs, resulting in significantly enhanced TE annotation.
Limitations
The two main components of the strategy are the reconstruction of ancestral genomes and the use of RepeatMasker for TE annotation. The first entails reconstructing ancestral genomes from up to 241 genomes; nevertheless, the accuracy of TE prediction at particular genomic loci may be affected by errors in the results. The main advantage of this method is that TEs in the SOI were called with the same method and threshold, ensuring the same confidence for TEs detected in RAGs and SOI. RepeatMasker applies a threshold to call elements.
Considering the frequency of mutations in modern genomes, reference-based TE annotation in RAGs provides a clear annotation of TEs, although it may possess drawbacks. These changes may not accurately represent the original sequences of the TEs, but they were crucial for the domestication of some TEs. Few “young” or less-diverged TEs were found in the RAGs, which could be explained by this. Rebuilding full-length TE progenitor sequences for several TE families, like in the case of L1, is necessary to enhance the quality of annotation in RAGs.
For effective analysis, the TE annotation approach employing freeware software necessitates a high-performance computing cluster. The computational cost of whole-genome TE annotation with RepeatMasker is high; for every genome, it takes 72 hours with 24 CPUs (1,728 CPU h), and this time varies with genome size.
Conclusion
This work proposes a novel approach that retains the widely used repository-based annotation method but adds information from several RAGs to produce a more comprehensive TE annotation. Given that matching sequences in RAGs are accessible with nucleotide-resolution alignment to the consensus sequences, this enables more sensitive recovery of degTEs and more interpretable results of newly annotated TEs. Since analogous sequences in RAGs are closer to the integrants’ ancestral state, the technique may also help update TE integrants that have already been annotated.
This work represents the first attempt to mine degTEs in an interest genome using entire RAGs. The method can be applied to many TE annotation techniques and species; however, precise degTE annotation would require high-quality RAGs. The L2 subfamilies showed the biggest increase in the increased annotation, each acquiring about 10 Mb degTEs, or 90%, from the present hg38 annotation.
There are many uses for the improved human TE annotation that results from the enhanced TE annotation approach, including figuring out how different TE-derived elements have changed over time and how certain TE subfamilies have spread throughout genomes. Building TE libraries de novo probing RAGs is another possible strategy for RAG-assisted enhanced TE annotation. This method may reveal hitherto unknown TE clades that have long since gone extinct and are very defective in many modern genomes. RAGs will improve in accuracy as more genomes are sequenced, and the method’s range of applications will be further expanded by the reconstruction of older RAGs.
Article Source: Reference Paper
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











 using a novel approach.){kind=link}