
The increasing relevance of protein design across a range of scientific fields drives the creation of integrative computational platforms that improve tool accessibility and interoperability. In order to do this, scientists present a web-based toolbox that expands upon the Damietta protein design engine by utilizing a framework for tensorized energy calculations. In addition to other tools like message-passing neural networks and molecular dynamics routines, the Damietta Server smoothly combines several design tools, enabling the user to conduct multiple operations on structural models and send them across tools. With an easy-to-use user interface, the toolkit may be applied to activities like conformational sampling, symmetric design, mutagenesis scanning, and core or interface design. The basic and applied biomedical research communities will benefit from the Damietta Server’s central resource for protein design and analysis, which will be made possible by the planned integration of additional technologies. There is no need to log in to access the toolbox.
Introduction
De novo protein design has quickly gained traction and established itself as a method for producing proteins with unique properties and uses. Moreover, directed evolution and other empirical engineering techniques are increasingly guided by computational protein design, which not only produces better outcomes but also explains the observed protein fitness. In order to achieve these goals, physics-based approaches are essential for defining sequence–structure connections, enabling the design of proteins down to the atomic level, and providing generalizable design frameworks. Furthermore, producing functional proteins is greatly aided by incorporating these techniques into specialized pipelines that include geometric patterns, statistical potentials, evolutionary profiles, and deep learning models. However, the biochemistry, biotechnology, and medical research communities are unable to apply physics-based methods more widely due to the specialized knowledge needed to set them up and integrate them with other instruments.
Understanding Damietta
Damietta, a framework developed in this work, presents a novel method for protein design computations. The system computes the interaction energy between two groups of atoms using a single tensor operation, in contrast to earlier approaches that assess interaction energies by looping over all interacting atom pairs. When used in the design, this one instruction, multiple data methodology may evaluate, compared to current methods, a substantially higher number of rotamers at atomistic precision. Moreover, a significant portion of the interaction data is precomputed and saved as 3D projections for the rotamer library, significantly decreasing runtime. Ultimately, multi-level parallelization is made possible by the constant-dimensional grids produced by this 3D tensorization. An energy function, molecular parameters, and a rotamer library generated from a single molecular mechanics force field form the foundation of the Damietta protein design framework. This has the dual benefit of not requiring any training process for parameterizing the terms in the scoring system and depending just on self-consistent terms. The researchers were able to create copper-binding and EGFR-inhibiting proteins with the Damietta design tools, as well as increase the affinity and adjust the activity of G-CSFR-binding designs.
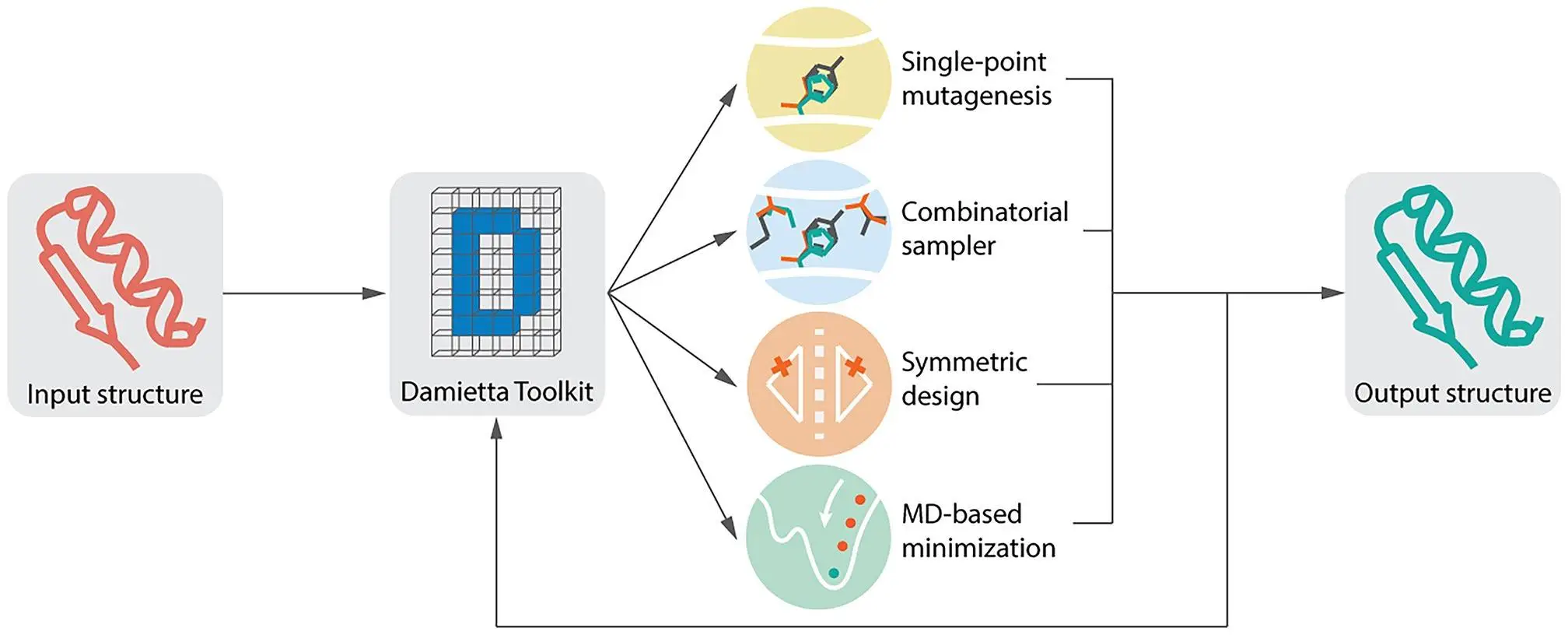
Aim and Key Features of The Damietta Server
The aim is to combine multiple protein modeling and design tools onto a single platform with an intuitive graphical user interface.
Researchers put into practice three Damietta applications:
- forced mutagenesis (sp)
- combinatorial (cs)
- symmetric (sd) design challenges.
- To attain complete freedom and enhance the caliber of the design models, researchers in these implementations additionally integrate the Damietta discrete rotamer samplers with molecular dynamics minimization techniques.
- Additionally, to assist users in evaluating the input and output of design activities, the Damietta Server is furnished with extra third-party tools.
- To create a position-specific mutation probability matrix, researchers specifically use message-passing neural networks in this version of the server.
- Researchers also use molecular dynamics methods for quality assessment and energy minimization of the final design models.
- Lastly, structure-centric flow, in which the input and output structure models may be easily passed across the various tools, is how the Damietta Server was designed with interoperability in mind.
- The Damietta Server is positioned as a complete design and modeling platform for various activities by virtue of these qualities.
Damietta Server Workflow
All operations on the Damietta Sever follow a three-stage procedure. The structure that begins and finishes the process allows the output structure to be passed on as input for a subsequent operation.
The structure is quickly pre-processed, analyzed, and shown during the workflow’s first stage. Pre-processing includes:
- Removing all non-protein atoms.
- Inserting hydrogens and other side chain atoms that are absent.
- Classifying each atom’s type in accordance with the CHARMM36 force field.
The mutational probabilities at each point in the backbone structure are then examined, as predicted by ProteinMPNN. The process produces two position-specific mutation probability matrices that are linked to the structure. The two probability matrices come from weights of neural networks that are solubility-enhancing and standard. The pre-processed input structure and sequence and input fields for the simulation type and parameters are graphically shown at the end of the first stage.
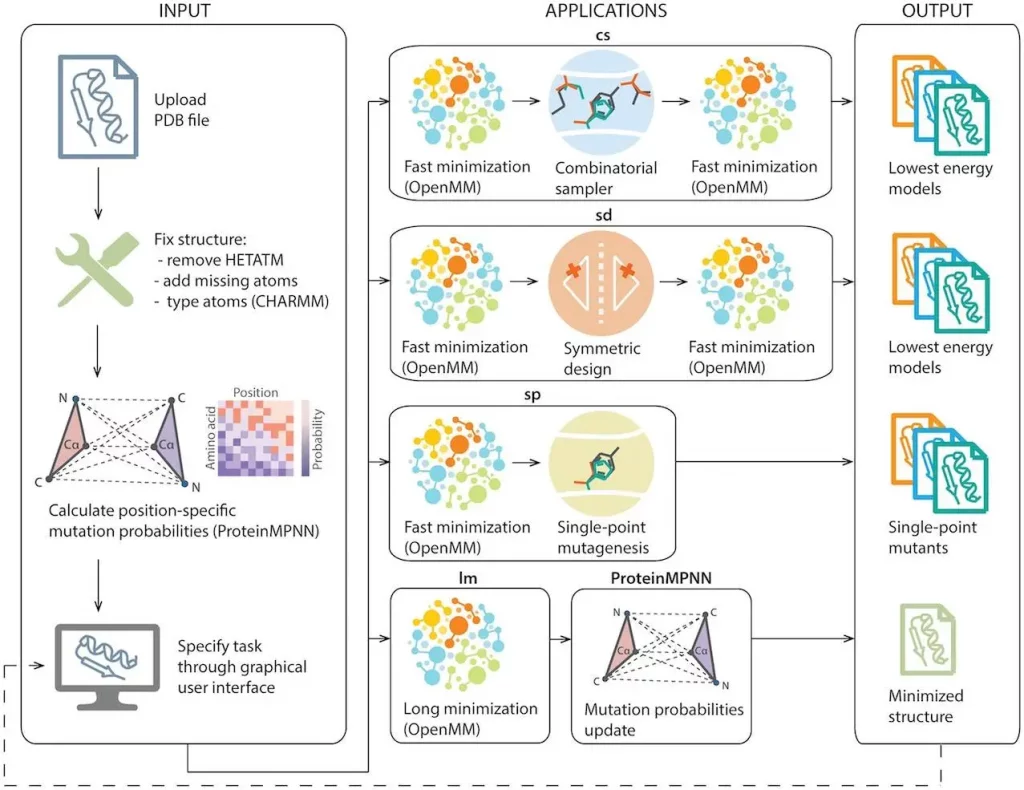
Repackaging and modifying existing side chains in amino acid sites requires the use of Damietta design tools. These tools remove side chain atoms, leaving behind atoms that describe an incoming rotamer’s host environment. While mutagenesis assesses side chain conformations of all target amino acid types, repacking assesses various wild-type side chain conformations for their internal and interaction energies with the host protein environment. The tools carry out all-atom minimization dynamics, repacking, and mutagenesis operations in different sampling orders and generate a set of lowest-energy models. The lengthy minimization tool generates a single model that represents the last frame of the simulation after performing all-atom minimization dynamics. Users can launch multiple runs with varied settings and tools simultaneously, and the output models can be utilized as inputs for more runs of the Damietta protein design toolkit.
Future Work
The long-term goal for development is to increase the range of applications that can be accessed via the Damietta Server. More specifically, researchers intend to include methods for local protein-protein docking, multi-state design, and tempering molecular dynamics routines on the sampling side. Researchers intend to enable the membrane environment to be set up on the scoring side.
Additionally, researchers intend to increase the range of supported chemotypes, including non-standard amino acids, metabolite compounds, and modified and tautomeric states of proteinogenic amino acids. LigandMPNN for sequence prediction and CGenFF-derived parameters for physical modeling can be used in tandem to expand this chemical space on the Damietta platform.
Conclusion
The Damietta Server is a protein design tool that provides a special set of tools, such as a first principles energy function and a rotamer library developed from molecular dynamics. Non-rotameric degrees of freedom and other conformational distortions can be sampled using this unbiased rotamer library, which spans a wider conformational space. Additionally, under the same force field, the server permits the extension of a rotamer library to encompass non-standard amino acid chemotypes. Scalable cloud computing resources for this study are provided by the German Network for Bioinformatics Infrastructure. Users may forward structures across different design tools with the Damietta Server, which smoothly integrates several design and analysis tools into a unified platform. The user-friendly graphical interface of the platform makes it simple for users to see their results.
Article Source: Reference Paper | The toolkit is available with no login requirement through https://damietta.de/.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}