
The field of biological sequence analysis has lately benefited from the revolutionary changes brought about by the development of self-supervised deep language models for natural language processing tasks. Conventional models show significant effectiveness in a variety of applications. These models are mostly based on the Transformer and BERT architectures. However, the quadratic computational complexity O(L2) of the attention mechanism places inherent limitations on these models, limiting their processing time and efficiency. The researchers from the Tokyo Institute of Technology introduce ProtHyena, a unique method that makes use of the Hyena operator, in order to address these restrictions. ProtHyena overcomes attention processes to reduce time complexity and enables the modeling of extra-long protein sequences down to the single amino acid level. This novel approach uses only 10% of the parameters usually needed by attention-based models to attain state-of-the-art results. The architecture of ProtHyena offers a highly effective method for training protein predictors, paving the way for the quick and effective analysis of biological sequences.
Introduction
Proteins are necessary for a variety of cellular functions, including metabolic activities and the maintenance of cell form by structural proteins. Comprehending proteins is essential to comprehending human biology and wellness, highlighting the necessity of sophisticated protein representation modeling employing machine learning methodologies. A major obstacle persists in getting relevant annotations for these sequences, even with the exponential increase of protein databases: most of them lack structural and functional annotations. Effective analysis techniques are required to make the most of the abundance of unlabeled protein sequences.
The Protein Language Model Landscape
Protein sequence representation learning has been transformed by deep learning models known as transformer models. These models learn universal embeddings through pre-training on millions of unlabeled protein sequences and then subsequently fine-tune them for particular protein applications. This methodology has been used in a number of fields, including biology, language, vision, and audio. Unfortunately, these models’ contextual capability is severely limited, and their application to larger sequences is hampered by the quadratic computing cost associated with the length of the input sequences.
Transformer-based models must employ techniques like linearized, low-rank, and sparse approximations due to their computing needs. While expressivity and processing speed are compromised, these strategies lessen the computational strain. Protein sequence inputs can have up to 2,048 tokens processed by hybrid models such as Protein RoBERTa using Longformer. Performer is used by foundation models such as scBERT and MolFormer to analyze gene expression data, while MolFormer adds linear attention to improve spatial interactions between chemical atoms.
Protein sequence representation using byte pair encoding (BPE) is limited, and protein models—which frequently have over 100 million parameters—present difficulties. BPE aggregates meaningful protein units, much like tokenizing words in language processing, although it has limits when it comes to single amino acid alterations. Since individual amino acids are physical analogs with substantial effects, their roles are crucial. Proteins demand a sophisticated strategy that resolves the specifics of individual amino acids while preserving a sense of the larger context. In order to effectively simulate these intricate biological sequences, this poses a unique problem in the field of proteins, beyond the limits of existing machine learning models and requiring creative solutions.
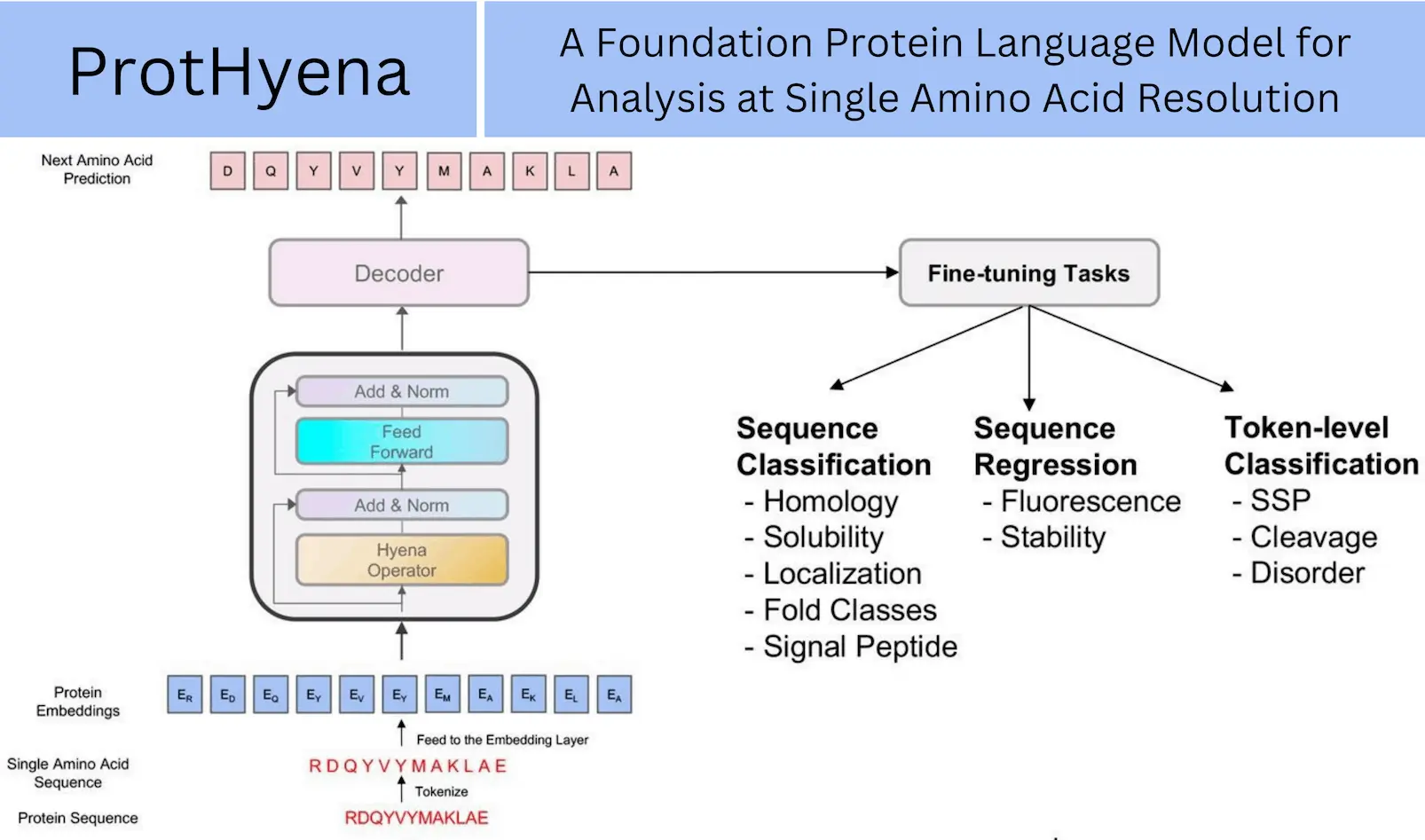
Understanding the Need for ProtHyena
Machine learning has evolved greatly with the recent development of Hyena, a big language model based on implicit convolutions, which reduces computational time complexity and allows the analysis of longer contextual sequences. As a result, ProtHyena—a quick and parameter-efficient foundation model that uses the Hyena operator to analyze protein data- was developed. Using attention-based methods, ProtHyena is able to capture genuine protein sequences at both long-range and single amino acid resolution. ProtHyena’s performance was evaluated by pre-training the model on the Pfam dataset and fine-tuning it over ten different downstream protein-related tasks. The outcomes show that in certain tasks, ProtHyena performs on par with or better than state-of-the-art performance. The databases can now be accessed by the general public, allowing researchers to assess the potential of newly developed protein sequence analysis methods in the future.

Breaking Down ProtHyena’s Capabilities
- ProtHyena outperforms attention-based models when it comes to protein modeling:
When it came to protein modeling, ProtHyena is a model built on the Hyena Operator that performed better than attention-based techniques. The same Pfam dataset was used to train two more models: ProtGPTtiny and the larger ProtGPT-base. The two models with the lowest confusion showed improved performance. This demonstrates how effectively ProtHyena models proteins, especially when dealing with the intricacies of protein sequences. ProtHyena’s reduced perplexity shows the model’s superior design and capability in this domain, especially when compared to the larger ProtGPT base.
- ProtHyena outperforms Transformers in modeling lengthy protein sequences:
ProtHyena offers significant speed and the ability to model longer sequences compared to attention-based models. At a sequence length of 1M, it provides speedups of up to 60 times by using a fused CUDA kernel to perform FFTConv operations. At sequence lengths greater than 2048, the Hyena operator performs better than traditional attention, while flash attention becomes noticeably slower. However, since it uses less hardware than flash-attention, the speed benefit becomes more noticeable for longer sequences. ProtHyena overcomes the limitations of earlier techniques by handling protein sequences up to one million amino acids in length. This allows it to solve issues such as information loss from sequence truncation and constraints imposed by larger vocabularies. This discovery broadens the scope of protein sequence analysis and contributes to a deeper comprehension of the structures and functions of proteins.
At longer sequence lengths, ProtHyena beats standard models by up to 60 times, showcasing its better design and optimized architecture in balancing computational load and performance, especially at crossing points between traditional attention and flash attention.
- ProtHyena performance on downstream tasks:
ProtHyena, which can be used to generate token- and sequence-level predictions, was tested against the BERT Transformer and TAPE Transformer, among other LSTM models.
Conclusion
This paper presented ProtHyena, which combines the Hyena operator to solve the computational challenges met by attention-based challenges. ProtHyena processes long protein sequences efficiently and delivers, and may sometimes outperform, state-of-the-art performance in many downstream tasks. ProteHyena, with the help of a few additional parameters, can proceed to make a significant leap in the field of protein analysis. The thorough pre-training and fine-tuning on the large Pfam dataset across ten different tasks shows the accuracy of the model. ProtHyena has more capabilities to level up, ensuring promising future advancements in the field.
Article Source: Reference Paper
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}