
Compound-protein interaction (CPI) prediction accuracy is crucial for drug discovery. Creating deep learning models that can be applied broadly requires expanding the CPI data through experimental validation. With their strong generalization performance in a range of tasks involving substances and proteins, large language models (LLMs) such as chemical and protein language models have become foundational models. This inspired researchers at Kyoto University to develop ChemGLaM, a chemical-genomics language model for predicting interactions between compounds and proteins. An interaction block based on two separate language models—MoLFormer for chemicals and ESM-2 for proteins—is used by the CPI prediction model ChemGLaM. Compared to previous models, this one predicts compound-protein interactions more accurately since it has been optimized for CPI datasets. The amalgamation of various models yields an intricate portrayal of interactions between compounds and proteins, supplying comprehensible perspectives into the workings of these interactions. This work emphasizes how independent foundation models can be integrated for multi-modality challenges.
Introduction
Drug discovery relies heavily on CPI prediction because experimental validation is expensive and time-consuming. Though they require accurate 3D protein structures and are computationally expensive, computational techniques like molecular docking and molecular dynamics simulations can predict CPIs. Using 2D compound structures and protein sequences, deep learning (DL) has become a powerful method for CPI prediction by merging genomic and chemical space. DL models can now extract representations of 2D chemical structures and protein sequences from internal or external CPI data thanks to DL algorithms, including transformer architectures, graph neural networks, and convolutional neural networks.
Promising progress has been made in resolving the data scarcity issue in CPI prediction by self-supervised learning. However, the scarcity of CPI data frequently places a constraint on the generalization performance of DL models. For example, BindingDB, one of the largest CPI datasets, has only a few million examples, whereas Davis and PDBbind have limited samples. When refined for downstream tasks, self-supervised learning, an unsupervised technique, can improve the generalization performance of models by learning representation from large-scale unlabeled datasets. Protein language models (PLMs) like ESM-2 and ProtTrans are useful for tasks involving protein sequences, but chemical language models (CLMs) like ChemBERTa and MoLFormer have demonstrated efficacy in the prediction of molecular properties.
Introducing ChemGLaM
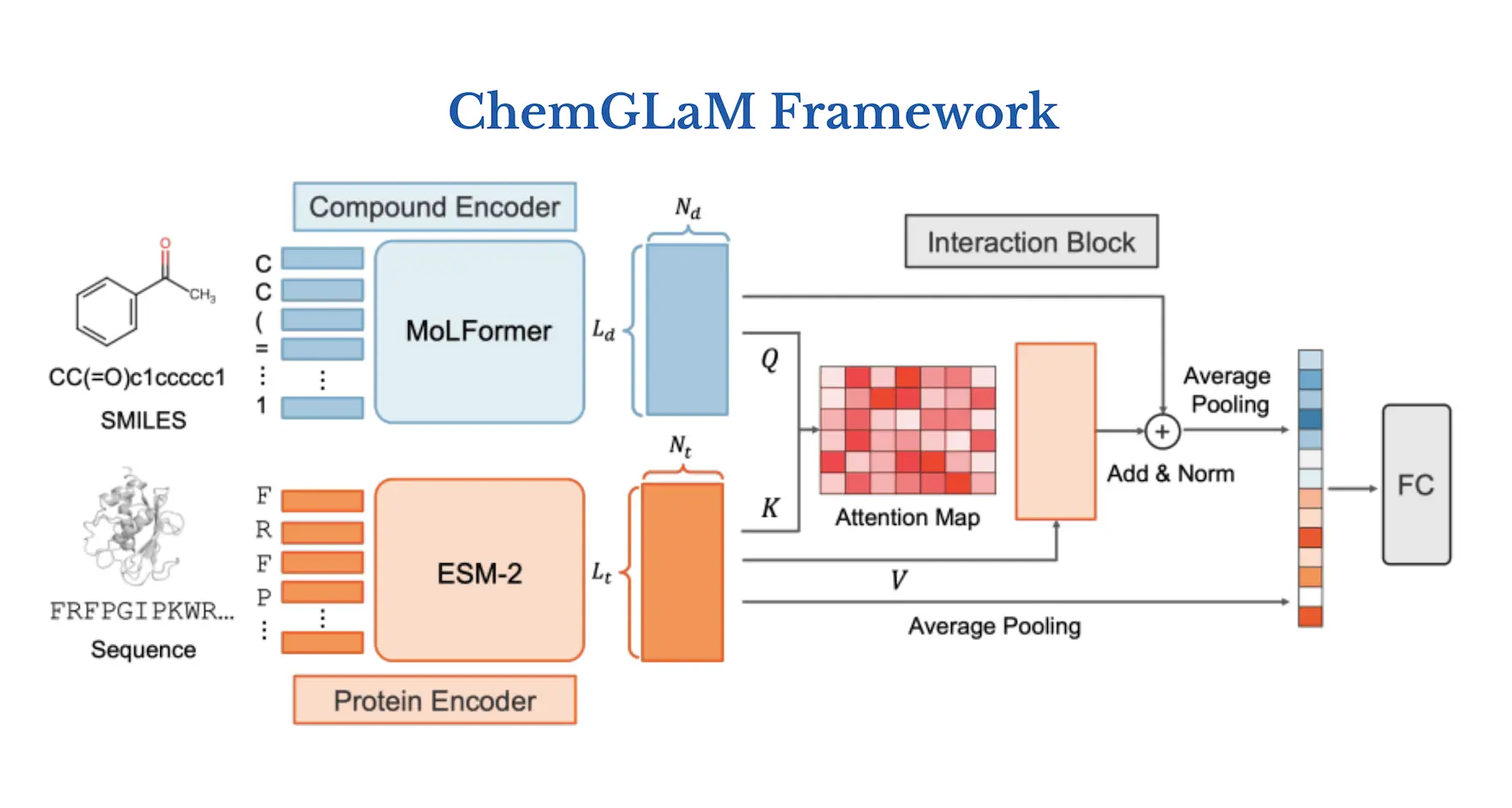
ChemGLaM, a chemical-genomics language model, is proposed by researchers to anticipate compound-protein interactions. The Interaction Block integrates the embeddings of the Compound Encoder and Protein Encoder, which are obtained from MoLFormer and ESM-2 with pre-trained model parameters, respectively, and employ cross-attention techniques. With the exception of the Protein Encoder, which was adjusted using CPI data from Davis, PDBbind, Metz, and BindingDB, the data was fine-tuned by the researchers. The suggested model shows that for multi-modality tasks like CPI prediction, integrating separately trained chemical and protein language models works well. Moreover, explaining the mechanism of compound-protein interaction can be facilitated by visualizing the learned cross-attention map.
Understanding the Architecture of ChemGLaM
ChemGLaM architecture consists of three parts:
(1) Compound Encoder: MoLFormer
Using training sequences of 1.1 billion unlabelled compounds from the PubChem and ZINC datasets, a simplified molecular input line entry system (SMILES) is used to teach MoLFormer, a large-scale chemical language model, how to represent molecules. The largest version of MoLFormer, called MoLFormer-XL, performs better in a variety of prediction tasks than the most advanced chemical foundation models. Since the whole model isn’t available in the public repository, researchers used the MoLFormer pre-trained with 10% of the MoLFormer-XL training dataset.
(2) Protein Encoder: ESM-2
The state-of-the-art protein language model Evolutionary Scale Modelling version 2 (ESM-2) is utilized for many different tasks, including predicting the structure and function of proteins. As a Protein Encoder of the ChemGLaM, the researchers selected the pre-trained ESM-2-T36-3B-UR50D, which is composed of 36 transformer blocks with 3B parameters learned from UniRef50.
(3) Interaction Block
The two aforementioned encoders are used by Interaction Block to learn compound-protein interactions. The input for the interaction block is the two outputs of the compound embeddings Hc = (Lc, Nc) and the protein embedding Hp = (Lp, Np), where Nc = 768 and Np = 2560 are the dimensions of the embeddings, and Lc and Lp are the token sizes of a compound SMILES and a protein sequence. Interaction Block uses multi-head cross-attention methods to update the compound’s representation once it has these embeddings.
Features of ChemGLaM
On the BindingDB dataset, the ChemGLaM model outperformed previous models in measures including accuracy, F1 score, area under the precision-recall curve (AUPRC), and area under the receiver operating characteristic curve (AUROC). The model also showed remarkable performance in zero-shot predictions. With an AUROC of 0.828, an AUPRC of 0.827, and an accuracy of 0.755, the model outperformed the others, demonstrating how well large-scale pre-training improved ChemGLaM’s generalization abilities. With an F1 score of 0.756, the model demonstrated exceptional performance and a high success percentage for positive classifications.
With the lowest Root Mean Square Error (RMSE) of 1.530 (±0.069), the highest Pearson correlation of 0.598 (±0.039), the highest Spearman correlation of 0.598 (±0.042), and the lowest MAE of 1.201 (±0.045), ChemGLaM—a novel chemical-genomics-based method—performed better in affinity prediction for the PDBbind dataset. This performance was contrasted with traditional techniques such as TransformerCPI and GraphDTA, which showed worse generalization performance. The Metz dataset provided more evidence of ChemGLaM’s generalizability in affinity prediction, as it surpassed baseline models in terms of RMSE, Pearson correlation coefficient, and Spearman correlation coefficient.
Relationship Between Protein Language Models’ Performance and Prediction Accuracy
This work investigates the relationship between the predictive power of protein language models (PLMs) and compound-protein interactions (CPIs). A ChemGLaM model was trained with a Protein Encoder based on PLMs from work by Lin et al. utilizing the PDBbind dataset. The findings demonstrated that models that perform better—that is, have lower validation perplexity and higher scores in important benchmarks—generally produce CPI estimates that are more accurate. Better measurements, such as lower root mean square error (RMSE) and greater Pearson correlation coefficients, corroborate this. On the PDBbind dataset, the ESM-2 (3B) model, which performed the best in the Lin et al. study, also produced the best CPI prediction results.
Application of ChemGLaM
ChemGLaM is a model that looks at the connections between protein sequences and compound SMILES using attention weights. It was applied to the reconstruction of attention weights for the PDBbind dataset in order to visualize CPIs. The model was utilized to illustrate the intricate relationship between glutathione disulfide (GSSG) and glutathione transporter (GshT). The hydrophobic pocket that forms between the N- and C-terminal lobes of GshT and is stabilized by hydrogen bonds, hydrophobic interactions, and van der Waals contacts is where GSSG attaches, according to the experimental structure. The highlighted areas matched the observed pattern of contact and were located in close proximity to each other. The complex building process is significantly influenced by hydrogen bonding interactions between the carbonyl groups in GSSH and Thr12 and Thr119 in GshT. The visualization indicated that only compound SMILES and protein sequences provided ChemGLaM with information about protein folding and binding events, with Val115 and the hydrophobic pocket being highlighted.
Conclusion
ChemGLaM is a new chemical-genomics language model that predicts CPIs by fusing the features of pretrained chemical and protein language models. Compared to other DL-based methods, this model’s multi-head cross-attention mechanism allows it to integrate the representations of chemicals and proteins with notable efficacy. Because ChemGLaM can use large-scale chemical and protein datasets for individual pre-training, it performs better on a variety of datasets, such as Davis, PDBbind, Metz, and BindingDB. Explainable insights into compound-protein interactions are provided by the model’s usage of visualized cross-attention maps. ChemGLaM results point to the need for more research and development in this area, with foundation models for chemicals and proteins being developed to improve CPI prediction abilities.
Article source: Reference Paper
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}