
Researchers from the Peter MacCallum Cancer Centre, Australia, have introduced an integrated open-source toolset called BARtab and bartools to streamline the analysis of cellular barcoding experiments. Cellular barcoding couples heritable DNA barcodes to high-throughput sequencing to enable precise tracing of cell lineages across conditions. The new tools automate the computational processing of barcode datasets, quality control, filtering, and quantification. Bartools additionally enable user-friendly visualization and interpretation of results. The tools support bulk DNA sequencing experiments and novel single-cell and spatial transcriptomics assays that read RNA barcodes. Compared to previous platforms, the BARtab and bartools workflow provides a scalable, portable, and interactive solution for handling diverse cellular barcoding data types. Lead developers highlight the tools’ capacity to democratize lineage tracing methods for widespread adoption. They plan future enhancements supporting additional barcode variants and integrations with common single-cell analysis packages.
Introducing Barcodes
Modern cellular barcoding techniques assign genetic barcodes to individual cells, enabling precise tracing of clonal lineages. These methods contribute to mapping developmental trajectories and lineage links among organisms and experimental systems, enabling the study of clonal dynamics at unprecedented scales.
Fundamentally, cellular barcodes are heritable during cell division, meaning that a unique barcode is engineered into each cell’s genome to generate a clonal lineage by allowing each daughter cell to acquire the same barcode as its parent. For lineage tracing research, synthetic barcodes made from genomic DNA can be employed. A high-throughput sequencing device can be used to sequence these barcodes after they have been isolated by PCR.
Synthetic barcodes can now be read out into single-cell transcriptomic datasets thanks to advancements in single-cell sequencing methods. These barcodes can be read out utilizing polyA capture-based single-cell RNA-seq techniques. They are present on mature mRNA transcripts and are cloned into reporter gene cassettes. This idea also applies to spatial transcriptomics technology based on sequencing.
Importance of Cellular Barcoding
A potent method for precisely tracking clonal lineages in a range of biological settings is cellular barcoding. This method traces cell lineages at the population or single-cell level using high-throughput sequencing and heritable synthetic barcodes. It has uses in immunology, developmental biology, and cancer research. Nevertheless, cellular barcoding dataset preprocessing and visualization are now lacking standardized and scalable open-source tools. By offering a scalable and portable pipeline for barcode extraction, quality assurance, filtering, and enumeration from high-throughput sequencing data, BARtab, and bartools seek to address this. The broad use of cellular barcoding in genomics research and the advancement of our knowledge of cellular lineage and function is made possible by these technologies, which also facilitate innovative expressed cellular barcoding approaches.
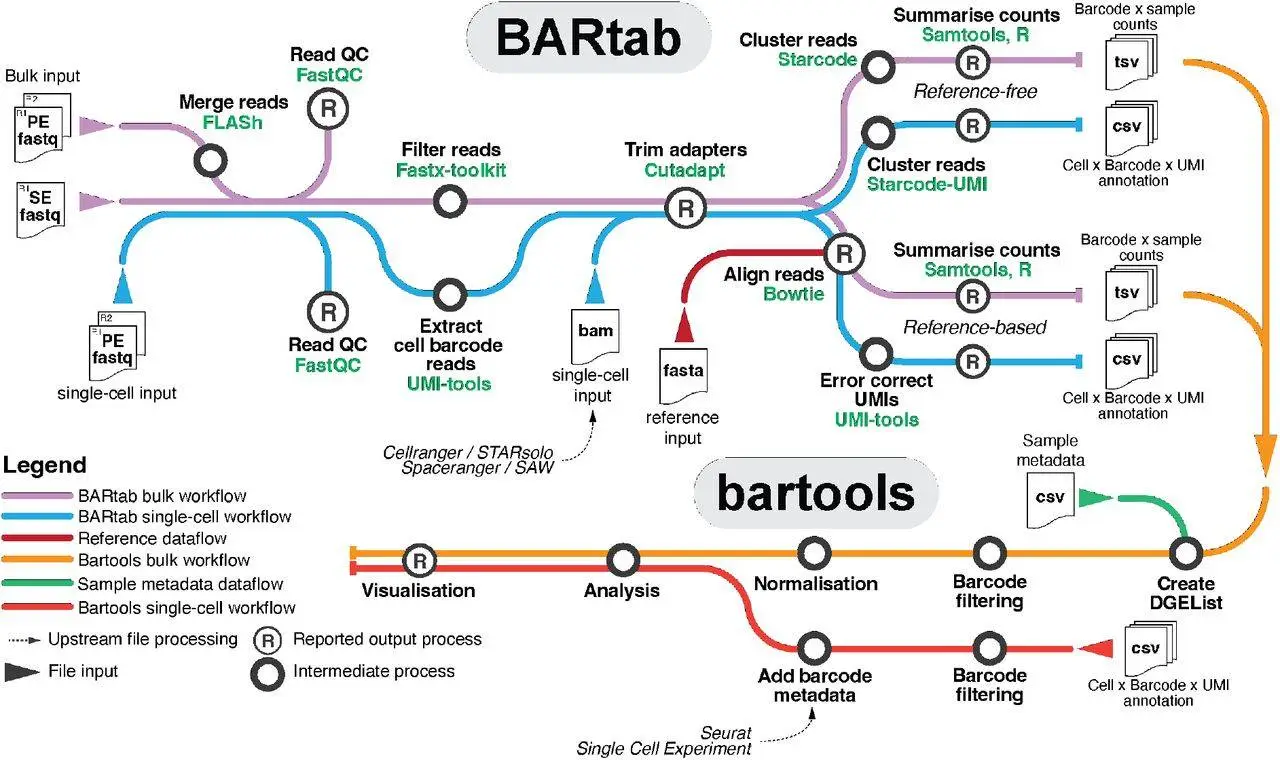
BARtab and bartools
Bartab and bartools are Nextflow pipeline and R packages designed to analyze synthetic cellular barcodes in the genome and transcriptome. These tools are an attempt to fill the gap in the open-source toolkit for processing and visualizing cellular barcoding datasets at the population and single-cell levels, which is the lack of scalable and standardized tools.
BARtab automates upstream procedures by offering a scalable and portable pipeline for barcode extraction, quality assurance, filtering, and enumeration from high-throughput sequencing data.
A variety of tools for analyzing and visualizing cellular barcoding datasets are available with the R package Bartools. From single-cell RNA-seq and spatial transcriptomics investigations, these tools include methods for retrieving and annotating transcribed barcodes. These technologies will aid in the widespread adoption of genomics research and advance our knowledge of cellular lineage and function by providing a standardized and user-friendly way for cellular barcoding.
Bartools and BARtab provide a standardized and user-friendly method for cellular barcoding in genomics research. They make it possible to analyze a variety of datasets, such as single-cell, bulk, and spatial transcriptomics data. A scalable workflow for barcode extraction, quality assurance, filtering, and enumeration from high-throughput sequencing data is provided by BARtab and bartools. They also cover techniques for taking transcribed barcodes from spatial transcriptomics and single-cell RNA-seq experiments and annotating them.
These approaches can further extend our understanding of cellular lineage and function by facilitating the widespread use of cellular barcoding in genomics research and opening up new applications in a range of biological contexts.
A standardized approach to extracting, annotating, and visualizing transcribed barcodes from single-cell RNA-seq and spatial transcriptomics experiments is provided by BARtab and bartools. This makes it more extensively applicable in the field of genomics by adding to the analytical toolkit for innovative cellular barcoding approaches. Bartools and BARtab can enable new applications in various biological contexts, thus improving our understanding of the origins and functions of cells.
Potentially strong toolkits like BARtab and bartools can handle a variety of cellular barcoding datasets. They are applied to the analysis of bulk, single-cell, and spatial transcriptomics data and are contrasted with current toolkits. The combined workflow of bartools and BARtab presented in the study highlights their portability and scalability, making them perfect for working with various datasets.
Workflow
The mice with leukemia cells MLL-AF9 were used in this study. These cells were divided into distinct treatment groups after being transduced with a lentivirus barcode. IBET-151, AraC and vehicle control (DMSO) were the treatment arms. IBET-151/AraC or vehicle control was applied in increasing quantities to the grown cells. Barcode sequencing was carried out at various intervals.
Technical replicates were included in the study, and correlation was evaluated to ensure data validity. Samples with correlation values below a rational cutoff were eliminated, while those with over 90% correlation were averaged. The CPM transformation was used to normalize the dataset, determining the number of barcodes in each remaining sample.
Conclusion
BARtab and bartools is a statistical tool that allows for additional analysis and interpretation of population-level cellular barcoding data by providing information on the abundance of each barcode across several samples.
Cellular barcoding approaches are increasingly utilized in biological research due to the increasing availability of techniques that combine clonal lineage with other cellular modalities, such as chromatin accessibility, cell surface protein expression, and spatial transcriptomics/epigenomics. These advanced techniques require robust data analysis tools that can be easily adapted to suit various experimental approaches and barcoding systems. The combined workflow provided by BARtab and bartools addresses these challenges by integrating pre-processing, quality control, and visualization of cellular barcoding datasets into a flexible workflow. Future developments will further enhance the bartools and BARtab framework to support additional lineage tracing strategies, such as CRISPR evolving barcodes, Cre-Lox recombination, and novel spatial technologies, thereby enhancing the potential of combining lineage information with single-cell and spatial genomics.
Article Source: Reference Paper | BARtab and bartools are both freely available at https://github.com/DaneVass/bartools and https://github.com/DaneVass/BARtab under MIT and GPL3 licenses respectively.
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}
[…] BARtab and Bartools: Revolutionizing Cellular Barcoding Analysis in Genomics Research […]