
Deep learning models like RosettaFold and AlphaFold2 can predict protein structure with high accuracy; however, they still have difficulties when it comes to intricate connections between large proteins. To combat this problem, scientists from The Hebrew University of Jerusalem introduced CombFold, a hierarchical and combinatorial assembly technique that uses pairwise interactions between subunits predicted by AlphaFold2 to predict the structures of large protein complexes. In this paper, researchers showed that CombFold accurately predicted 72% of complexes in two datasets of 60 large, asymmetric assemblies, with a 20% higher structural coverage than Protein Data Bank entries. High-confidence predictions were obtained when the approach was applied to complexes from Complex Portal that lacked structure but had known stoichiometry. CombFold is a promising method for extending structural coverage beyond monomeric proteins since it allows for the incorporation of distance constraints and the quick enumeration of potential complex stoichiometries.
Understanding Methods for Determination of Protein Assemblies
In cells, the majority of proteins function as multimolecular assemblies. A protein typically has a few dozen interactions. Important tasks like energy transduction4, transport, and signal transduction are carried out by these assemblies. Determining the three-dimensional structures of these assemblies is essential for comprehending their evolution and function, analyzing the consequences of mutations, and possibly even for use in drug discovery. X-ray crystallography and nuclear magnetic resonance spectroscopy are two common structural characterization methods that face difficulties due to the huge size of some assemblies and conformational variability. Cryo-electron microscopy (cryo-EM) has made strides, but big assembly structure determination at high throughput remains a difficult task.
Growing Importance of AI on Protein Structure Prediction
The capacity to predict high-accuracy protein structures has significantly improved recently thanks to deep learning approaches. The release of AlphaFold2 and RosettaFold was one of the most significant developments. Although AlphaFold2 was created to predict single-chain proteins, it can also use the same architecture to predict protein complexes. Soon after AlphaFold2 was released, several methods were created to use it to predict multichain protein complexes: initially, by offsetting the residue index and then by utilizing a linker. Similar methods were applied to train AlphaFold-Multimer (AFM), a program that uses paired and padded multiple sequence alignment to predict multimeric complexes with great accuracy. AFM achieves a success rate of 40–70% on many pairwise protein-protein docking benchmarks for complexes with two to nine chains up to 1,536 in total length.
Challenges in Structure Prediction
The AFM application for predicting structures of large assemblies faces several challenges. The first is the requirement for significant resources, like a GPU with a sizable memory capacity. Currently, common GPUs have about 20GB of memory, which allows complexes up to 1,800 and 3,000 amino acids to be predicted for AFM versions 2.2 and 2.3, respectively. However, this limits many researchers’ practical capacity to forecast large-sized structures as AFM memory utilization grows with the number of amino acids. As the number of chains and amino acids rises, sampling with a lot of constraints becomes more challenging, which hinders the model’s ability to converge on precise structures. Large, multimolecular complex prediction is an out-of-domain inference setup for AFM, as it was trained only on cropped regions. Finally, AFM converges to a single structure, making it challenging to obtain a diverse set of predictions for the same target.
The two main types of approaches used to assemble multiprotein complexes are docking-based methods and integrative modeling methods based on experimental data. In order to compute models, the first category is integrative modeling, which uses data from several sources, including small-angle X-ray scattering, cryo-EM, co-evolution, and FRET. With the use of specialist software, this data is transformed into geographical constraints and integrated into an integrative modeling methodology. Four iterations comprise the workflow: data collection, scoring, sampling, and model validation. Global data-driven optimization algorithms, including genetic or Monte Carlo algorithms, are frequently used for the sampling of candidate models. Large and diverse systems, like the ~52 MDa nuclear pore complex, can benefit from the application of integrative structural modeling. Recently developed AlphaLink allows sampling with distance constraints using AlphaFold2.
Second is Pairwise docking, which is a technique for complicated prediction that does not require additional input data, but because there are a lot of docked configurations, high accuracy is hard to accomplish. Using a genetic algorithm-driven stochastic search, Multi-LZerD assembles molecules into multimolecular assemblies. By adding a single subunit to the subassembly in each iteration, Kuzu et al. build multimolecular complexes iteratively. Using pairwise docking configurations between subunits, the hierarchical and combinatorial CombDock approach creates subassemblies of two or more subunits and connects them. Reaching high accuracy in multisubunit docking is challenging, nevertheless, because pairwise interaction scoring and docking methods have low accuracy. Therefore, it is essential to consider a larger number of pairwise protein-protein docking models in the multiprotein assembly stage.
Understanding CombFold
Inspired by the work of the recently developed MoLPC method, which relies on AlphaFold2 to produce configurations for pairs and triplets of chains, the authors of this paper developed CombFold by combining AlphaFold2 with a deterministic combinatorial assembly algorithm. CombFold is a new assembly technique that maximizes the likelihood of correctly building complexes by using a limited number of AlphaFold2 interactions. The technique achieved a top-1 success rate of 62% and a top-10 success rate of 72% on two benchmarks of large heteromeric assemblies. In comparison to experimental structures, CombFold also yields a 20% improvement in structural coverage. In addition, the technique delivers a 57% top-1 success rate on homomeric complexes employed in MoLPC validation efforts. With more than 3,000 amino acids, the approach assembles six of the seven CASP15 targets with success.
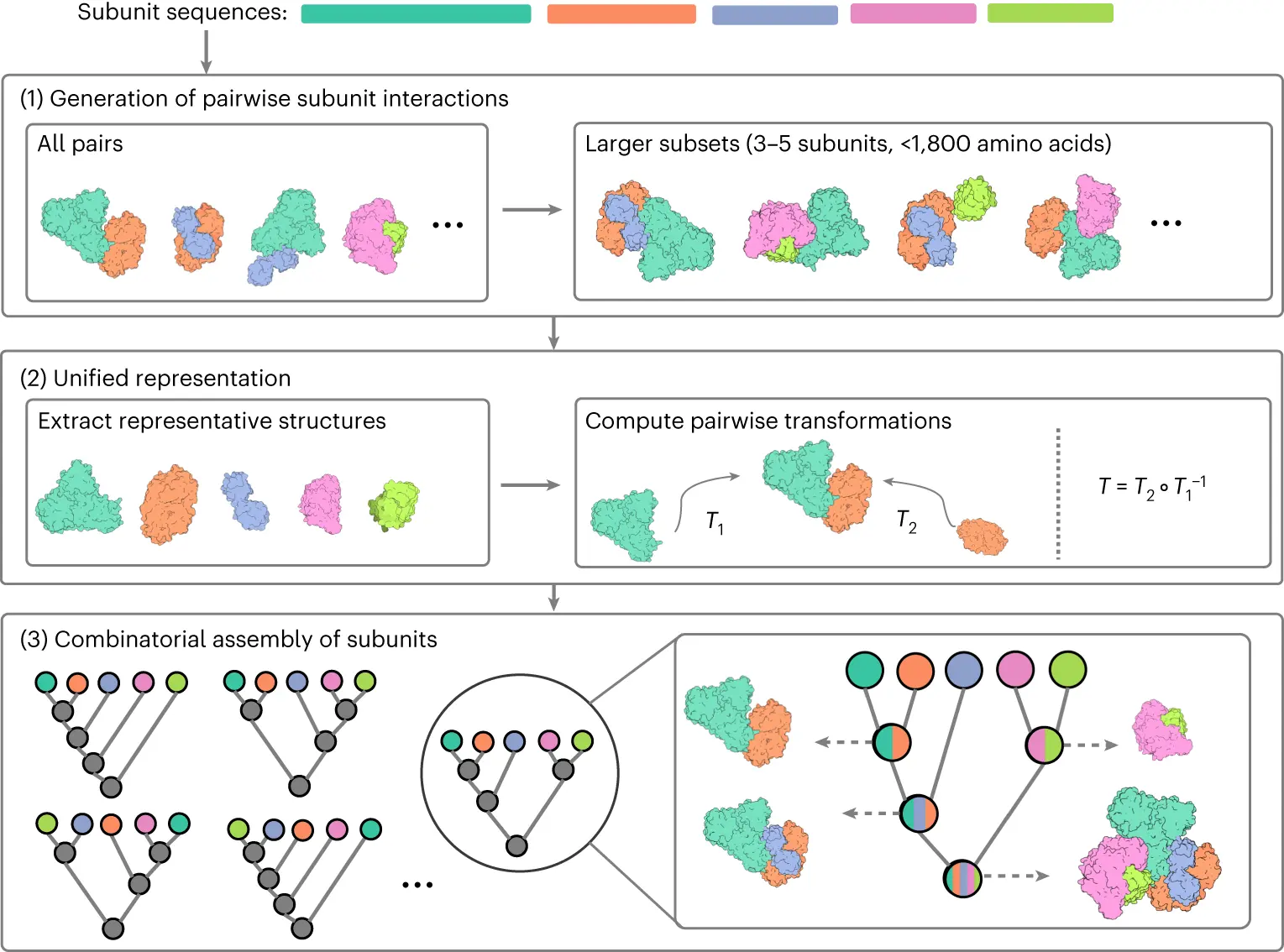
Using complexes from Complex Portal that have a known stoichiometry but an unknown structure, the approach has produced predictions that may be trusted. Subunit sequences and optional distance constraints are fed into CombFold, and the result is a collection of constructed structures. A subunit may consist of a domain or a single chain. The method relies on paired interactions to achieve hierarchical and combinatorial assembly. Since the complex can be broken into subunits that are appropriate for the GPU memory limit, there is, in theory, no limit to the size of the complex. The current implementation supports up to 128 subunits. Three main phases comprise the operation of CombFold:
- AFM-generated pairwise subunit interactions
- Unified subunit and interaction representation construction
- Combinatorial subunit assembly
Limitations
The ability of AFM sampling to generate paired subunit interactions limits its success rate. This method can be further improved by collecting more pairwise orientations from pairwise docking methods and improving AFM sampling by enabling dropout during inference. Some complexes are dynamic and exist in numerous states, whilst others assemble into stable structures. The heterogeneity can be conformational with flexible proteins or compositional with subunits that interact momentarily or a mix of the two. It is challenging to deal with this variability. For instance, counting compositions during assembly can be a similar strategy to address compositional variability as stoichiometry. Currently, conformational heterogeneity is addressed using additional structural data from mass spectrometry, cryo-electron tomography, cryo-EM, and single-molecule FRET.
Conclusion
CombFold is a method that uses AFM substructures for pairs or larger subsets of input subunits to predict the structure of large multisubunit protein complexes. Accurately predicted assemblies are obtained by counting the top-scoring assembly trees using the combinatorial assembly algorithm. For increased accuracy, the technique can incorporate data from crosslinked mass spectrometry datasets. With a top-10 success rate of 57-74% for both homomeric and heteromeric assemblies, CombFold has been verified on four datasets. CombFold can be used to solve structures that might not completely cover sequences since it can increase the structural coverage of huge complexes that have been solved experimentally by 20%. CombFold makes use of crosslinking mass spectrometry and massive databases of protein-protein interactions and assemblies from Complex Portal, Corum, and STRING. The main obstacle in using assembly methods is the building of complexes without known stoichiometry. Researchers plan to extend the method to enumerate stoichiometries so assembly can be done more effectively.
Article source: Reference Paper | The code, Colab notebook, and tutorial for CombFold are available on GitHub | The Code Ocean capsule for running the assembly algorithm is available at https://codeocean.com/capsule/8791899
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}