
The geometric characteristics of protein-nucleic acid complexes, which are explored using structural models, are crucial for proteins to recognize and bind nucleic acids. However, these features are limited when compared to unbound proteins. PNAbind is a deep learning method that the University of Southern California researchers have developed for predicting protein-nucleic acid (protein-NA) binding based on a protein’s apo structure. The research demonstrates that graph neural network-based models can predict the geometric characteristics of protein molecular surfaces, which in turn can predict RNA binding. These models enable a better understanding of the chemical and structural characteristics determining recognition by differentiating between specificity for DNA and RNA binding. At the individual residue level, the models also predict where nucleic acid (NA) binding sites would be found. The predictions show the promise of these models in understanding protein structures, as they are evaluated against benchmark datasets and match experimental RNA binding data.
Introduction
In biomolecular activities such as transcription, translation, regulation, and genome structural organization, protein structure is essential. Target recognition and binding by NA binding proteins (NABP) is dependent on the geometric, chemical, and electrostatic features of solvent-exposed side chains as well as their spatial arrangement. Although they are challenging to produce experimentally, structural models of protein-NA complexes offer insights into the physical mechanisms underpinning protein-NA recognition. Just 23% of the roughly 44,000 protein structures with known NA binding functions that are currently in the protein data bank include protein complexes bound to NA. Large-scale projected protein structures that may be examined directly using computational techniques are now available thanks to developments in protein structure prediction.
The physical properties of binding surfaces of protein-NA (PNA) complexes were discovered to be shared by NABP in the early days of structural biology. These features included significantly positive electrostatic potential, propensity for particular residue-nucleotide combinations to form hydrogen bonds, and enrichment of polar and positively charged sidechains. As a result, computer methods for predicting NA binding based on protein sequence and structure were developed; these early efforts have been highlighted in a number of reviews and comparison studies.
It has proven possible to predict protein function and binding, including NA binding, using graph neural networks (GNNs). Protein graph representations can be created using DeepFRI, GraphSite, and GeoBind, which rely on sequence embedding models and Ca-Ca atom distances. Protein residues are represented as nodes in the GeoBind approach, which uses a GNN to create a dense graph inside a sliding sphere. Each node is allocated sequence and structural properties, and residue-level predictions of DNA and RNA binding sites are learned by latent spatial encoding. ScanNet predicts protein-protein binding interfaces by using a point-cloud model of a protein and learning an embedding of nearby atoms and residues. GeoBind predicts DNA and RNA binding sites using quasi-geodesic convolutions over point clouds, while PeSTo employs a geometric transformer to predict protein-protein interfaces. While closely related, other techniques like MaSIF and PST-PRNA also use GNNs.
Understanding PNAbind
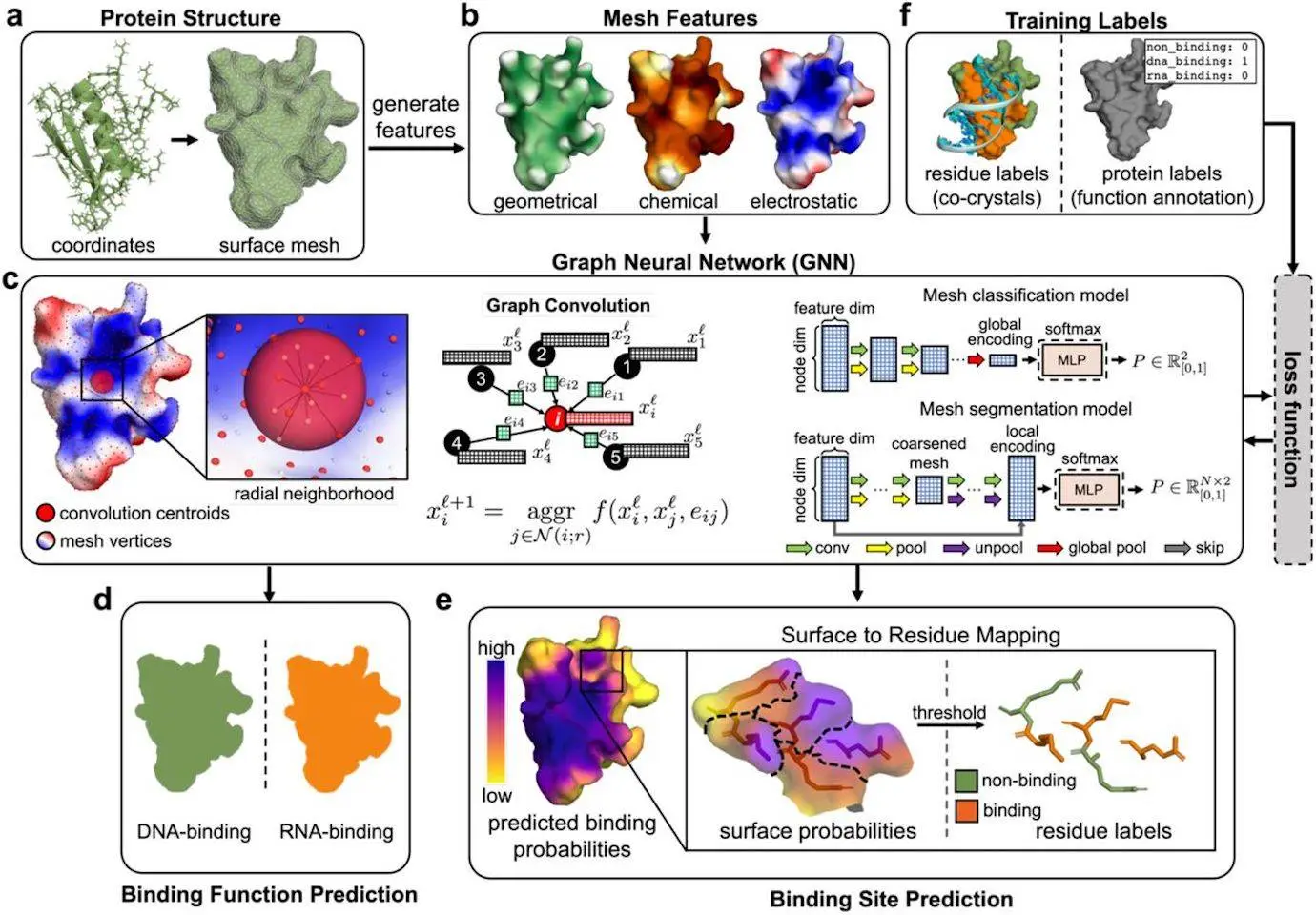
PNAbind is a method based on graph neural networks (GNN) for predicting DNA and RNA binding functions as well as binding sites using protein structures. In order to capture crucial geometric properties of a protein precisely, such as binding pockets and the complementary form of the binding site with a binding target, the method uses a mesh discretization of the solvent-excluded molecular surface. Features of the protein structure are mapped to the surface, including geometric, electrostatic, and chemical properties. For better binding site prediction, additional sequence alignments providing evolutionary information can also be included. In order to anticipate NA binding, the GNN models first learn spatial arrangement encodings.
The method works with proteins that differ greatly in terms of size, biological activity, and structural fold. These models are able to discriminate between DNA and RNA binding, predicting DNA and RNA binding with excellent accuracy. The approach generates low false positive rates on a negative control dataset and generalizes to both bound and unbound protein structures. For the deoxycytidine deaminase APOBEC3G, which is essential for the restriction of the HIV-1 virus, the predictions agree with experimental RNA binding evidence. PNAbind has several uses, including locating unidentified NA binding proteins, determining functional binding residues, improving the interpretation of biochemical data, and offering background knowledge for protein-NA complex modeling.
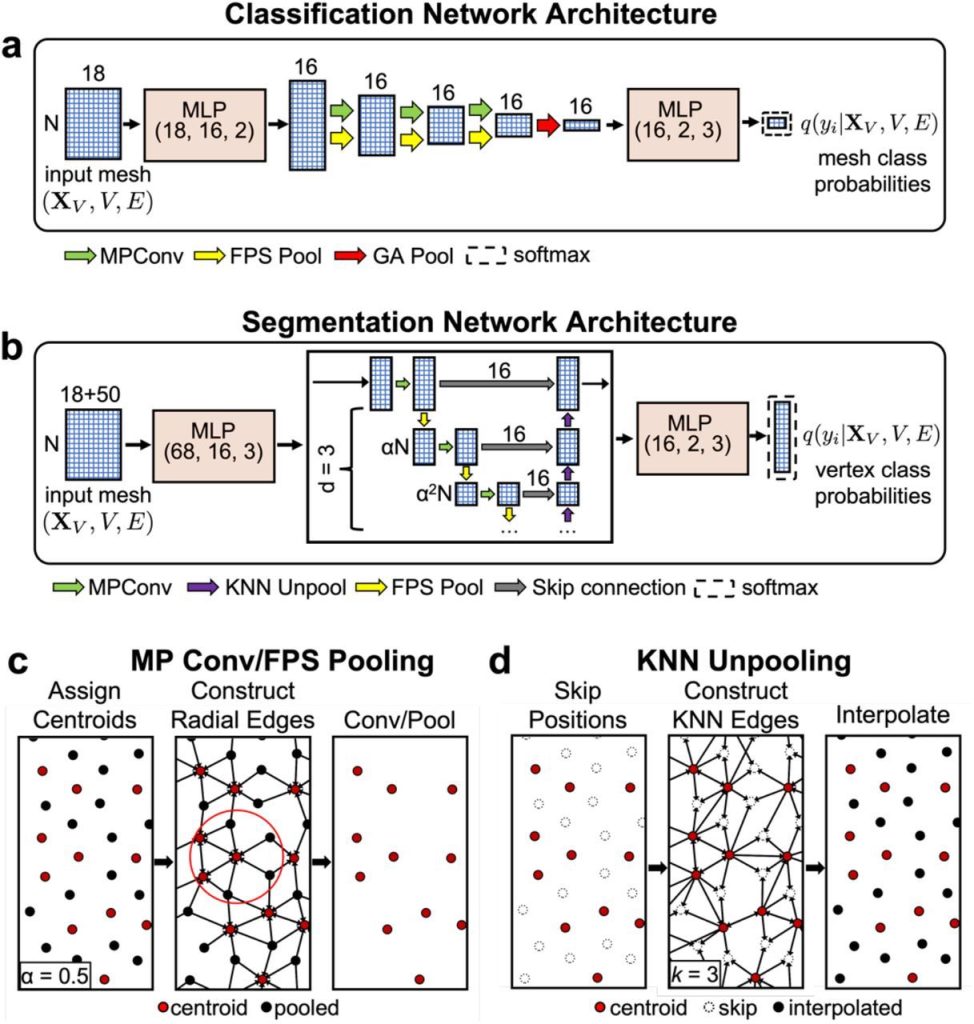
Image Source: https://doi.org/10.1101/2024.02.27.582387
Applications of PNAbind
PNAbind models use the geometric, chemical, and electrostatic features of protein molecule surfaces to predict binding. These models estimate the overall binding function and classify proteins using GNNs, a flexible neural network architecture. Using graph segmentation, they also predict where NA binding sites would be found on the surface of proteins. The models are a useful tool for protein analysis since they respect the permutation symmetry of graphs and are invariant to all isometries of Euclidean space.
Building Block of PNAbind model
Graph convolution, pooling and unpooling layers, and multilayer perceptron (MLP) modules are the primary building blocks of the models. Learned functions called graph convolutions are performed across a graph’s edges to collect data on the local graph topology. The convolution kernel is dependent on edge features, which characterize the features of neighboring vertices as well as distance and angle correlations, and the convolutions are carried out over edges determined by a radial distance threshold. Spatial groupings of physicochemical and geometric information indicative of NA binding can be encoded by performing the convolution repeatedly and aggregating the activations of the kernel function over the edges of vertex neighborhoods. Pooling is a technique that gradually removes some vertices from the mesh to coarsen it, which permits the radius of the subsequent convolutional layer to be increased without causing the number of edges in the ensuing graph to rise in an approximately cubic manner. The model can learn encodings over a range of distance scales by adjusting the radius. Restoring the mesh to its initial resolution is the purpose of unpooling, which is solely required for binding site prediction. The output of the networks represents probability distributions that are produced per-mesh (vertex) for binding function (site) prediction after the final global (local) encodings are translated via an MLP module.
Conclusion
Using surface-based protein representations, PNAbind predicts global protein binding functions, such as DNA-binding versus RNA-binding. Protein molecular surface shape and chemistry associated with established NA recognition processes drive feature selection. The binding function of structural motifs inside the binding interface may be associated with specific geometries, such as binding elements that can be inserted into major or minor dsDNA grooves or binding pockets that can be used to capture nucleotide bases in ssDNA. The findings regarding the discrimination of binding functions between DNA and RNA emphasize the significance of geometric information, which outweighs chemical and electrostatic characteristics. With more than 200 million predicted protein structures available in the AlphaFold database, PNAbind can be used for high-throughput annotation of protein-NABPs. High-accuracy binding site prediction, identification of functionally significant residues, and pre-modeling information for protein-NA complexes are all made possible by this model. PNAbind is a structure-based method that can be used for protein-ligand binding sites or other functional annotations. It offers a broad means of describing the relationship between the biological function and structural characteristics of proteins, which can have an impact on any class of proteins.
Article source: Reference Paper | Data, source code, and documentation for implementing and training PNAbind models is available on GitHub
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











 binding based on a protein's apo structure.){kind=link}
Electrodynamics not electrostatics!!!!
Dear Dr.Safonkini Oleh,
Thank you for engaging with the article. The reference paper delves into electrostatic properties. You can find detailed insights in the reference pre-print https://doi.org/10.1101/2024.02.27.582387. Feel free to discuss if you have additional questions!
Team CBIRT