
Scaling laws predict the presence of over a trillion species on our planet. However, only a small fraction of them have been studied. Deep learning models applied for work in life sciences generally depend on the size and quality of training or reference datasets. The study suggests a data-centric strategy to improve deep learning models in biology by collaborating with stakeholders in biodiversity and nature parks on five continents, spanning half of the world’s biomes, and setting up a worldwide supply chain for biological data and metagenomics. In protein sequence analysis, MSA supplementation provides a data advantage that outperforms currently available public data in AlphaFold2’s protein folding problem inference. The model BaseFold proposed by the researchers exceeds the performance of traditional AlphaFold2. This improved the quality of the structures that can be used to get better results from docking.
Introduction
Over the past few years, there has been an increase in the number of deep learning models used for various biological tasks. Given its significance for structural biology and drug development, the protein folding problem is particularly well-known. AlphaFold2, RoseTTAFold, and ESMFold, for instance, are offering encouraging and frequently quite precise predictions for this challenge. A great deal of study and work has been done to improve the architecture of these models to increase performance. However, as these models rely on the structure and protein sequence datasets available for training, the researchers used a data-centric approach to improve the biology deep learning models, using the protein folding problem as an example for this study. Scaling laws indicate that the public sequence datasets for deep learning in biology, like MGnify, NCBI, and UniProt, constitute less than 0.000001% of all species on Earth. The majority of sequences that are entered into these databases come from model organisms, humans, and mammals that are raised in confined spaces in laboratories. Moreover, these services do not provide consistent geolocation and environmental metadata for sequence data gathered in an environmentally friendly manner.
The commercialization of biological samples requires consistent traceability and profit sharing, as stipulated by the Nagoya Protocol and the United Nations Convention on Biological Diversity (CBD). These laws provide ethical Access and Benefit Sharing (ABS) when biological resources are commercialized, guaranteeing that material examined for research objectives becomes surprisingly valuable commercial assets. Still, there have been cases in which research materials contributed to commercial assets of very high value without a reexamination of the initial agreements under which the samples were obtained. This has resulted in disputes over biopiracy and hindered the advancement of revolutionary resources for business and public health.
Understanding BaseFold
The study addresses the knowledge gap on genomic sequence diversity and proposes a global metagenomics and biological data supply chain with the goal of enhancing biological deep learning models through digital sequence information sharing. Six billion relationships are produced by the knowledge graph, which gathers information, protein, and genome sequences from metagenomic sampling trips. When different sequences are supplemented, models such as AlphaFold2 can perform better on orphan proteins with deep multiple sequence alignments (MSAs). When comparing projected structures to ground-truth crystal structures, the root mean squared deviation (RMSD) can be lowered by as much as 80%. Additionally, the enhanced docking performance and structure predictions for different competition targets are shown.
Addressing the knowledge gap of biological sequence diversity
Environmental samples were taken for the metagenomic data gathering process in 23 countries on 5 continents, which covered half of the world’s biomes. Six billion relationships were found in the knowledge network created from the sequencing sites. The goal of using MSA generation downstream for AlphaFold2 predictions was to guarantee that sequences came from long, high-quality (meta)genomic assemblies. The material of the existing sequence database, MGnify, is not lengthy enough to encompass whole open-reading frames for bigger proteins and is fragmented. A vast range of geological and chemical conditions, covering a temperature range of -9 to 99◦C (15.8 to 210◦F) and a pH range of 1 to 12, were sampled using the Basecamp Research Data (BRD) database. The diversity of protein sequences deposited in BRD was assessed by comparing the size of the databases when clustered at 90%, 50%, and 10%.
Enhancing AlphaFold2 with the addition of MSA
A metagenomic sequence dataset containing one billion sequences from CASP15 and CAMEO targets was supplemented with MSA to improve the pace of sequence searches. A 50% identity criterion was reached for MGnify and BRD clustering, guaranteeing the insertion of sequences without sacrificing speed. The goal of the biennial worldwide experiment CASP (Critical Assessment of Structure Prediction) is to improve the current state of the art in protein three-dimensional structure modeling based only on the amino acid sequence. Participants shared their guesses for proteins whose experimental structures have not yet been added to the Protein Data Bank.
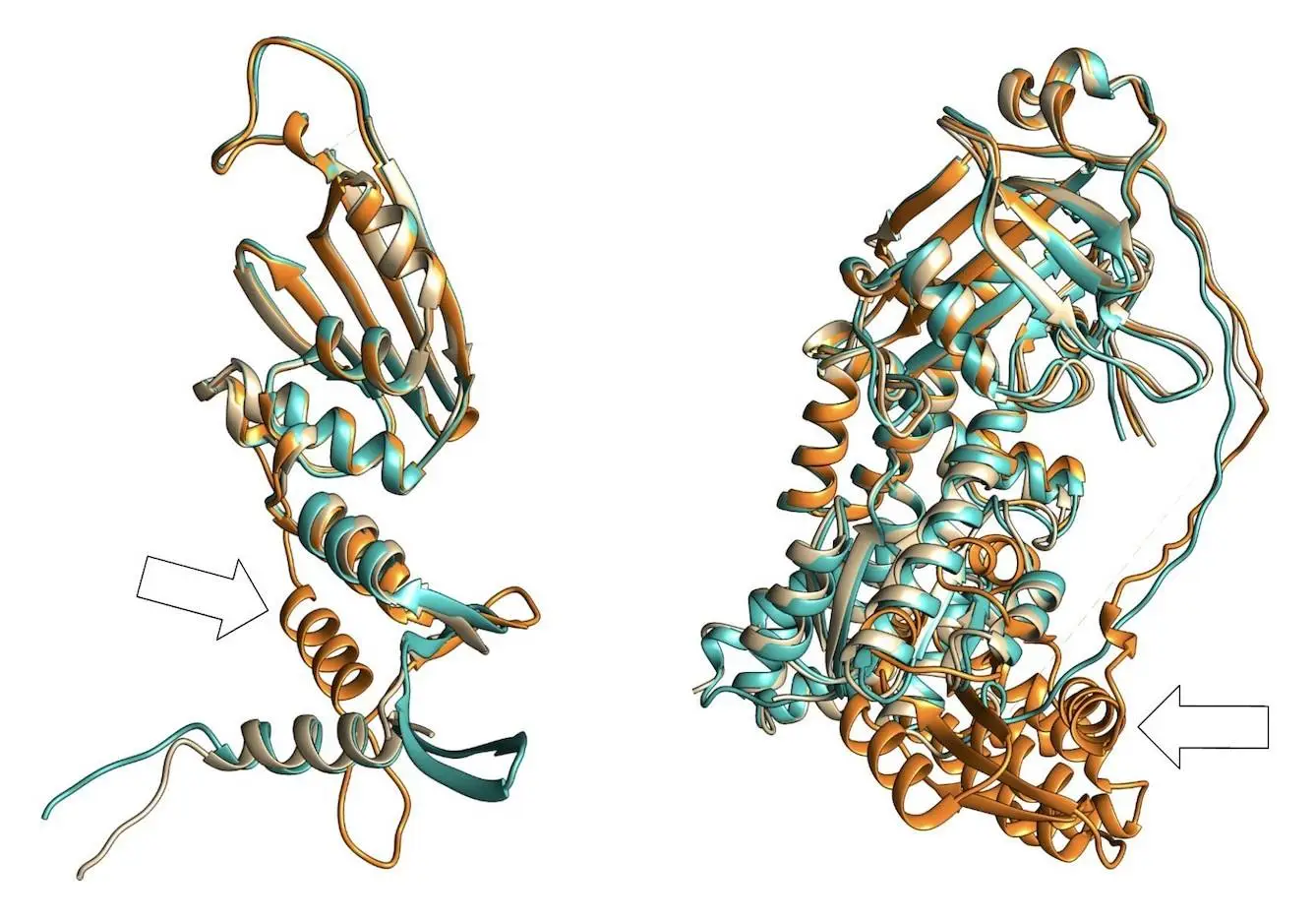
Predicted Local Distance Difference Test (pLDDT) scores were used to compare the structural predrudictions of 49 CASP15 regular targets, including single monomeric protein crystal structures. According to the study, improvements in pLDDT scores were observed in 61.22% of targets, with increases ranging from 0.08 to 24. For every target with a higher pLDDT score, the Root Mean Square Deviation (RMSD) score—a crucial statistic in CASP competitions—was determined. For targets with a higher pLDDT score, the SwissModel server showed an RMSD score reduction ranging from 0.02 to 3.33. The findings emphasize how crucial MSA supplementation is for raising pLDDT and lowering RMSD values.
Weekly updates on 3D protein prediction models that are ready to be deposited into the PDB are available on the web platform CAMEO (Continuous Automated Model Evaluation). Its main objective is to assess quality estimates for predictions of protein structure. In this study, 26 CAMEO targets were forecasted, and 57% showed an increase in pLDDT scores. For these targets, MSA supplementation decreased RMSD scores and increased pLDDT. The visualization of specific cases using structural superimposition demonstrates the platform’s attention to the changing needs of the structural bioinformatics community.
Conclusion
The diversity of life on Earth is a very small portion of the data available, including the space of genomic and protein sequences. A data supply chain is being developed in collaboration with biodiversity stakeholders to enhance deep learning models in biology. AlphaFold2 predictions can be much enhanced by adding a variety of sequences from this supply chain to MSAs; in fact, some RMSD values can drop by more than 80%. Moreover, this enhancement may enhance the efficacy of substrate/ligand docking, which may benefit efforts in drug discovery and enzyme engineering. More research is intended to examine how reference databases’ sequences are put together and how best to divide up sequence space so that accuracy and speed of inference are balanced. The objective of data collection that is in line with the United Nations ABS principles is to improve deep learning models in the life sciences by bringing biodiversity conservation activities together in a data-centric manner.
Article source: Reference Paper | Reference Article
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}