
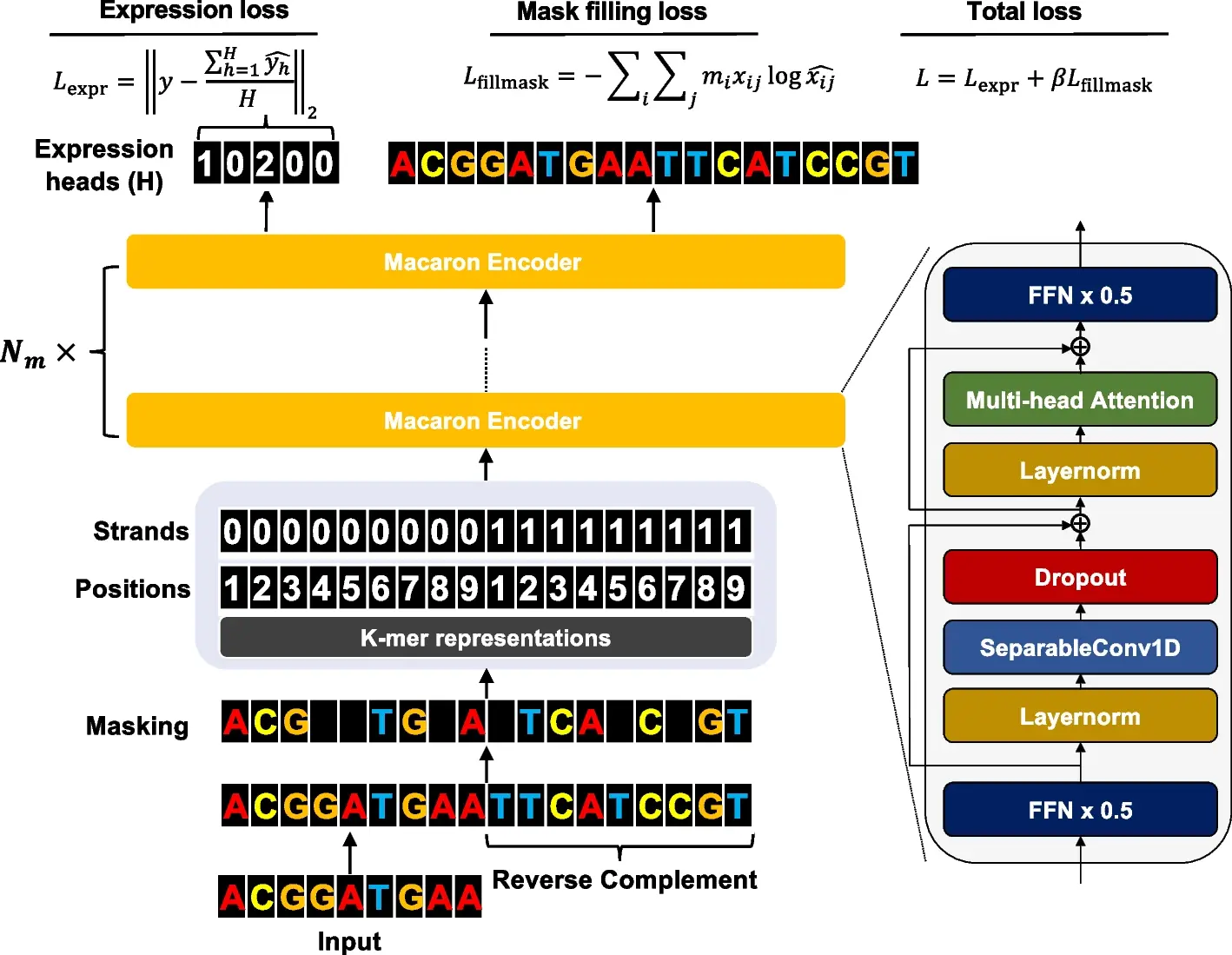
The ability to comprehensively decode the cis-regulatory logic that controls the expression values is made possible by the revolutionary high-throughput measurement of the cis-regulatory activity of millions of randomly produced promoters. Researchers from the University of Minnesota developed an end-to-end transformer encoder architecture called Performer to predict the expression values from DNA sequences. Proformer is a new learning technique that makes use of a Transformer encoder architecture like a Macaron, with two half-step feed-forward (FFN) layers positioned at the start and finish of each encoder block. After the initial FFN layer and before the multi-head attention layer, a separable 1D convolution layer is added. Along with learned positional and strand embeddings, the sliding k-mers from one-hot encoded sequences are mapped onto a continuous embedding. To keep transformer models from collapsing during training on tiny amounts of data, Proformer adds numerous expression heads with mask filling. The understanding of how cis-regulatory sequences determine expression values has improved as a result of this design’s notable improvement over traditional designs.
Understanding Gene Expression
A basic mechanism required for the coordinated operation of all living things is gene expression. One of the most important problems in molecular biology is predicting a gene’s expression level based on its promoter or enhancer sequences. Applications of this challenge range from comprehending gene regulation to biotechnologically altering gene expression. Large-scale and high-throughput massively parallel reporter assays (MPRAs), which allow the high-throughput investigation of gene expression and regulatory elements, have been essential in recent progress. Large-scale investigations into evolutionary reconstructions, variation effect prediction, and functional annotation of potential regulatory components are made possible by MPARs. For instance, the enhancer activity of tens of millions of distinct regions from the Drosophila genome was examined using STARR-seq. Numerous sequence tags and microarray analyses were utilized to assess hundreds of thousands of different mammalian promoter or enhancer variations.
Learning about different Machine Learning Methods
Large-scale DNA sequences, such as MPRA data, can have complex correlations and patterns that can be recognized using machine learning techniques. The following can be predicted through machine learning models: binding affinities, chromatin characteristics, DNA methylation, RBP (RNA-binding protein) binding, and gene expression levels using convolutional neural networks (CNN) and recurrent neural networks (RNN). To predict the function of DNA sequences, deep learning models like DeepSEA, DanQ, DeepATT, and CRMnet integrate a bi-directional long short-term memory network with 1D CNN. Using millions of random promoter sequence data, Vaishnav et al. developed a CNN- and Transformer-based deep learning model for gene expression level prediction. CRMnet is a deep learning model that combines 1D CNN, Transformer, and U-net architectures for sequence-to-expression prediction.
Introducing Proformer
In order to estimate expression values from millions of DNA sequences, the study created Proformer, an end-to-end transformer encoder architecture. The transformer uses multiple expression heads (MEH) to stabilize convergence. Proformer, which predicted gene expression using millions of randomly selected promoter sequences, came in third place in the DREAM challenge. In order to provide insights into creating and benchmarking genomic models across various contexts, the study assessed model architecture and training strategy choices on functional genomic region prediction. A new way to understand and describe how cis-regulatory sequences determine expression values is offered by the Proformer model.
Proformer Architecture: Overview
Proformer predicted expression values from promoter sequences using an encoder architecture akin to a Macaron. This encoder can be understood as a numerical solver of an Ordinary Differential Equation for a convection-diffusion equation in a multi-particle dynamic system since it has two half-step feed forward layers. Because input sequences are stochastic, it was hypothesized that the design would better recover connections between nucleotide patterns and expression levels. As in Transformer systems like Conformer, a separable 1D convolution layer was implemented to collect local signals. Every macaron block has residual connections to make gradients flow more naturally and avoid overfitting. The Macaron-like encoder underwent multiple iterations to guarantee precise predictions.
Prediction of chromatin accessibility by Proformer’s MEH with mask filling
Proformer was utilized to forecast ATAC-seq signals, a method that gauges chromatin accessibility throughout the genome using DNA sequences. Two head designs that applied to comparable situations were tested. When the model size was relatively small, the global average pooling performed well; nevertheless, over-parameterized models typically performed poorly. With a block size of four and a dimension size of one, MEH with mask filling produced the greatest results. The entire genome’s chromatin accessibility is measured using the ATAC-seq method. The Proformer’s application in this endeavor may aid in the prediction of the mean ATAC-seq signal from DNA sequences in comparable circumstances.
Importance of Proformer’s MEH with mask filling in improvement of prediction performance on hold-out validation data
Using a block size of 4 and a dimension size 512, the final DREAM challenge model was trained using 95% of the organisers’ data. At the checkpoint following the sixth epoch, the validation Pearson’s R was at its highest point. On the validation data, MEH with mask filling outperformed global average pooling in terms of Pearson’s R. Performance from the ablation trial was comparable to that of global average pooling with a single expression head. On hold-out validation data, MEH with GLU activation yielded higher unweighted Pearson’s R and Spearman’s Rho but lower weighted scores than global average pooling.
Better performance of Proformer over other methods involving random promoter dataset
A comparative analysis was conducted between the Proformer model and other models that are currently in use, such as CRMNet. The training set comprised 51,339,035 samples, and 2,954 random and 61,150 native yeast promoter sequences’ expression levels were included in the evaluation set. The training data was used to train the Proformer model, and Pearson’s R value was determined so that it could be compared to alternative techniques. R values of 0.971 for the native promoter dataset indicated that the Proformer and CRMNet models performed similarly, according to the results. With R values of 0.991, the Proformer model beat all other models on the random promoter dataset.
Conclusion
The Proformer architecture is a new approach to analyzing MPRA data and understanding regulatory logic. It uses multiple expression heads (MEH) with mask filling to prevent over-parameterized transformer models from collapsing when training on small amounts of data. This design strategy has shown better performance and stable convergence when applied to predicting expression values and chromatin accessibility from DNA sequences. This suggests that MEH with mask filling could be an effective design strategy for similar regression tasks in genomics, especially when using large, over-parameterized models. Future research should explore the development of a versatile pre-trained network for large-scale DNA data, similar to how language models are fine-tuned for specific tasks in natural language processing. This approach could be applied to genomics, utilizing transfer learning from existing DNA data, providing an exciting research topic.
Article source: Reference Paper | Codes for important analyses used in this study are available on GitHub
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}