
Despite the difficulties presented by the current methods depending solely on gene expression data, RNA sequencing data has been incorporated into Gene regulatory networks (GRN) inference methods, improving the capacity to learn complicated pathways from a few independent data points. Here, scientists offer LINGER (Lifelong neural network for gene regulation), a machine-learning technique that uses chromatin accessibility and single-cell paired gene expression data to infer GRNs. To estimate transcription factor activity from bulk or single-cell gene expression data, LINGER employs GRN inference from reference single-cell multiome data. This approach reveals a complex regulatory landscape of genome-wide association studies, improving accuracy above previous methods by a factor of four to seven. It makes use of the wealth of gene expression data that is currently accessible to pinpoint driving regulators from case-control research.
Introduction
Gene regulatory networks are groups of molecular regulators that combine to control gene expression and silencing in particular biological settings. To comprehend how cells carry out a variety of tasks, how they modify gene expression in response to environmental cues, and how noncoding genetic variations result in disease, a thorough grasp of gene regulation is essential. Transcription factors (TFs), which bind DNA regulatory regions to either activate or repress target gene expression, makeup GRNs. TF-TG trans-regulation from gene expression is the main challenge in the inference of GRNs. Chromin accessibility measures and co-expression-based techniques are helpful, but reliable inference is hampered by undirected edges and correlations. Although heterogeneity reduces the accuracy of inference, a statistical model called PECA was created to fit TG expression by TF expression and RE accessibility across various cell types.
The Advent of Single-cell Sequencing Technology
The development of single-cell sequencing technology has made it possible to do extremely precise regulatory research at the individual cell type level. Co-expression analysis, such as PIDC and SCENIC, allows for cell type-specific trans-regulation inference based on single-cell RNA sequencing (scRNA-seq) data. A transposase-accessible chromatin (scATAC-seq) single-cell sequencing assay can be used to infer trans-regulation, as demonstrated by DeepTFni. Numerous techniques combine unpaired scRNA-seq and scATAC-seq data to infer trans-regulation.
These techniques, which link TFs to REs through motif matching and link REs to TGs through the covariation of RE–TG or physical base pair distance, include IReNA, SOMatic, UnpairReg, CoupledNMF, DC3, and others. To transfer labels from scRNA-seq to scATAC-seq data, scJoint was recently created; this could lead to better cell GRN inference. Despite massive efforts, GRN inference accuracy has been surprisingly low, hardly outperforming random predictions. These issues can be addressed with the help of recent developments in single-cell sequencing, such as SCENIC+.
Nevertheless, three significant obstacles still exist in GRN inference.
- First, it is still difficult to understand such a complicated mechanism from sparse data. Single-cell data provides a lot of cells, but the majority of them are not independent.
- Secondly, it is hard to incorporate past knowledge into non-linear models, such as motif matching.
- Third, the experimental data-derived inferred GRN accuracy is slightly superior to random prediction.
Understanding LINGER
In this paper, researchers propose a technique known as LINGER (Lifelong neural network for gene regulation) to overcome these obstacles. This study makes several contributions to the field of GRN inference. To mitigate the problem of little data but extensive parameters, LINGER first employs lifelong learning, a previously stated concept that includes large-scale external bulk data. Second, LINGER incorporates previous knowledge into the model by integrating TF-RE motif-matching knowledge through manifold regularization. Third, LINGER’s accuracy is a four- to seven-fold relative improvement. Fourth, LINGER makes it possible to determine driving regulators and estimate TF activity only from gene expression data.
Uses of LINGER
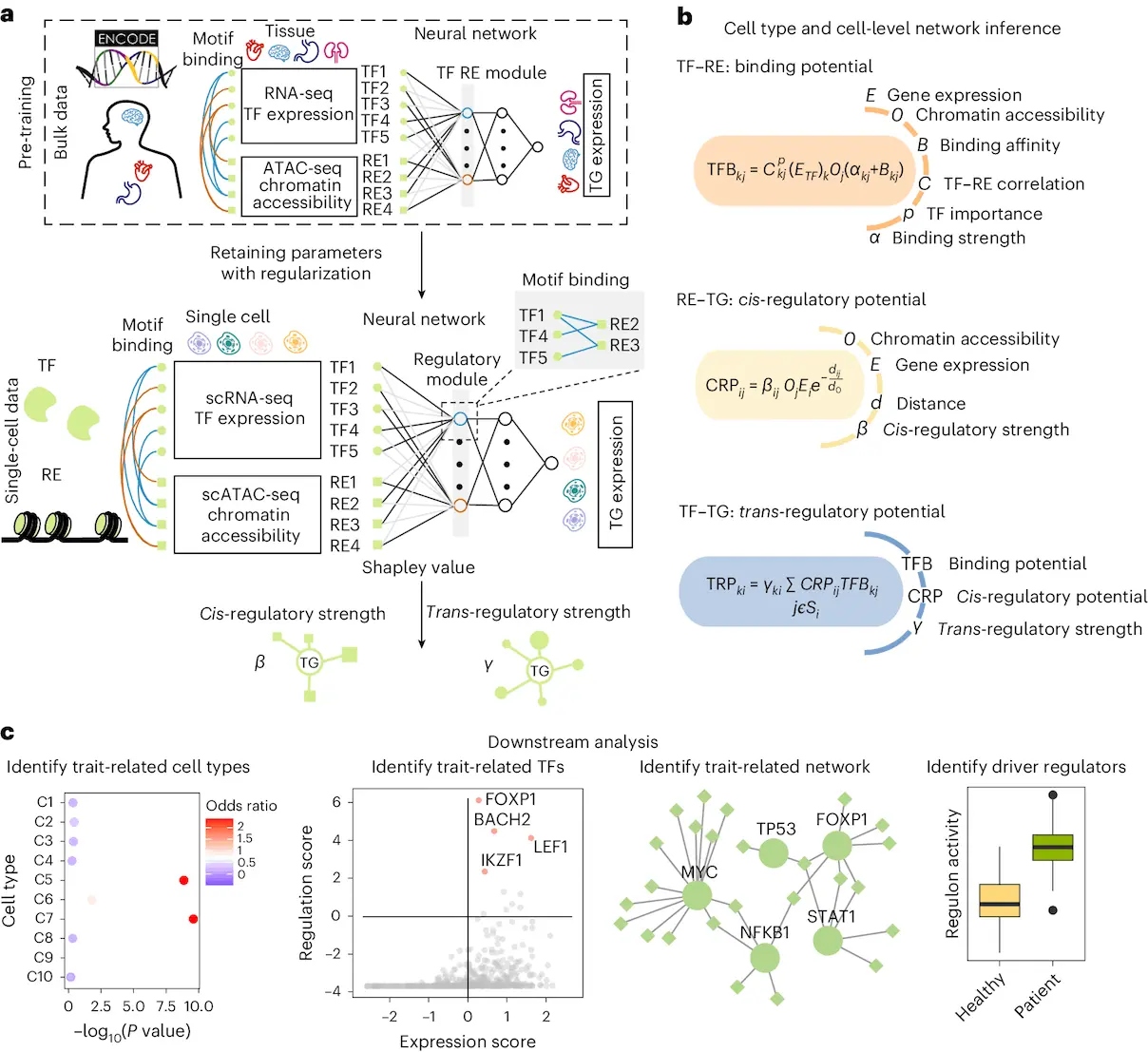
- LINGER was created to deduce GRNs from single-cell multiome data. It generates a cell population GRN, cell type-specific GRNs, and cell-level GRNs based on count matrices of gene expression and chromatin accessibility, as well as cell type annotation. Three different kinds of interactions are seen in each GRN: TF-binding (TF–RE), cis-regulation (RE–TG), and trans-regulation (TF–TG). It should be noted that while TF–TF interactions are taken into account in TF–TG couples, TF self-regulation—which is difficult to simulate in the absence of additional data—is not. The capacity of LINGER to incorporate the entire gene regulation profile from external bulk data sets it apart. Continuous learning, another name for lifelong machine learning, is how this is accomplished. The idea behind lifelong learning is that people can pick up new skills quickly and with little effort if they have prior expertise. It has been demonstrated that lifelong learning makes greater use of the knowledge gained from prior tasks to better understand the present one.
- Researchers used a public multiome dataset of peripheral blood mononuclear cells (PBMCs) from 10× Genomics to evaluate LINGER’s performance. They performed a comparison analysis between the two models to determine whether a linear model is sufficient for modeling gene expression or if a non-linear model is required. The first model predicts how TFs and REs express TG using an elastic net. The second model shares the design of LINGER and is a three-layer neural network called the single-cell neural network (scNN). The researchers used fivefold cross-validation to evaluate the two models’ capacity to predict gene expression.
- LINGER outperforms Pearson’s correlation coefficient (PCC) and motif binding affinity-based predictions for MYC binding locations in the H1 cell line. LINGER consistently shows the highest AUC and AUPR ratios for all 20 TFs in PBMCs, and its overall distributions are significantly greater than those of the other PBMCs (P ≤ 8.72 × 10−5). For H1 data, LINGER performs better than alternative techniques (P ≤ 6.68 × 10−6). Additionally, researchers contrasted LINGER with SCENIC+, a cutting-edge technique that predicts TF–RE couples using multiome single-cell data. Since SCENIC+ does not offer a continuous score for every RE, the accuracy was measured by the F1 score, according to the researchers. For all 20 TFs binding site predictions, LINGER performs better.
Conclusion
LINGER is a neural network-based technique that uses bulk datasets and TF–RE motif matching information to infer GRNs from paired single-cell multiomic data. LINGER achieves significantly higher GRN inference accuracy than current tools. Using lifetime machine learning to take advantage of various cellular settings and continuously update the model as fresh data come in is a significant innovation. This tackles longstanding issues caused by sparse single-cell datasets and large parameter spaces obstructing intricate model fitting. Pre-training on large collections is a benefit of LINGER’s lifetime learning approach, which enables users to quickly retrain the model for their research while utilizing publically accessible resources without direct access. When the model adjusts to the new single-cell data, the lifelong learning process will encourage it to retain previous information from the bulk data. There is a trade-off between fitting new data and holding onto past knowledge. When fitting the new data, there is no restriction on the range of prior knowledge variation. The amount of loss experienced throughout the fitting process of the new data determines how much the final conclusion deviates from the previous understanding. LINGER will automatically figure out how to maximize the use of the data from both datasets by making this trade-off.
Article Source: Reference Paper | The software is available under the GPLv3 license on GitHub and Zenodo.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}