
Deep learning-based virtual screening provides a more effective way to find molecules that resemble drugs, and virtual sources give chemists useful information. Leiden University researchers present PCMol, a multi-target model that trains the de novo generative model on target proteins using the latent embeddings extracted from AlphaFold. Protein descriptors are a useful tactic for expanding the quantitative structure-activity relation (QSAR) models’ prediction range and applicability. Using structural relationships between proteins, AlphaFold latent embeddings within a generative model for small molecules enable extrapolation on the target side based on similarities to other proteins and interpolation within the chemical space of known highly active compounds, which is especially relevant for understudied or novel targets.
Introduction
Drug development research is increasingly concentrating on computational methods to choose novel candidate molecules in silico. These substances have to interact chemically in a certain way with the binding site residues of protein targets. These interactions can be confirmed using in vitro bioactivity testing; however, physics-based computational techniques such as molecular docking or QSAR models are frequently performed prior to thorough virtual molecule screening. Choosing compounds for virtual screening entails either creating them from scratch using de novo molecule generation models or choosing them from huge libraries of synthesizable chemicals like Enamine Real.
Conventional molecule-generating methodologies, such as QSAR models and PCM models, are intended to produce compounds for a single target or a limited subset of homologous proteins. On the other hand, more modern techniques incorporate structural limitations depending on the aim, providing advantages comparable to PCM modeling. Certain models employ the target protein’s amino acid sequence for conditioning; however, in the absence of further pre-processing or modeling, this may not provide an adequate description of protein targets. Alternative methods concentrate on localized structural target representations, highlighting the protein’s active location. Even with these developments, there are still a lot of unanswered questions about how to describe target-based limitations, which makes this a fascinating field for study.
Large protein-language models such as RosettaFOLD2, ESM, and AlphaFold have been rapidly adopted, greatly advancing computational structural biology and chemistry. These models show their extensive, informative content by producing high-quality predictions based on big amino acid sequences and 3D protein backbone pairings. Reusing the high-quality protein representations these models have acquired for different tasks makes sense, considering the financial and computing resources needed for training or recreating these models. The viability of employing these embeddings as inputs for various deep learning models, such as molecular docking, protein binding site prediction, conserved or variant viral protein residue prediction, and protein homology inference, has been investigated in recent publications.
Role of Generative Models on Drug Design
In drug design, generative models are essential because they can propose new chemical structures that meet particular restrictions on molecular properties while also taking advantage of the active compound space found in ligand bioactivity datasets such as Pyrus or CHEMBL. To score and rank the produced compounds, these models frequently make use of generative machine learning approaches, such as Quantitative Structure-Activity Relation (QSAR) models. These models represent compounds and calculate binding affinities to particular target proteins using numerical descriptors. Through methods like drug-target interaction modeling (DTI) and proteochemometrics (PCM), they can be expanded by adding target protein information. This makes it possible to anticipate the bioactivity of a ligand on a variety of protein targets, which may help to identify ligands with high selectivity or off-target effects. This also enables the use of larger training datasets, a desirable feature in deep learning applications.
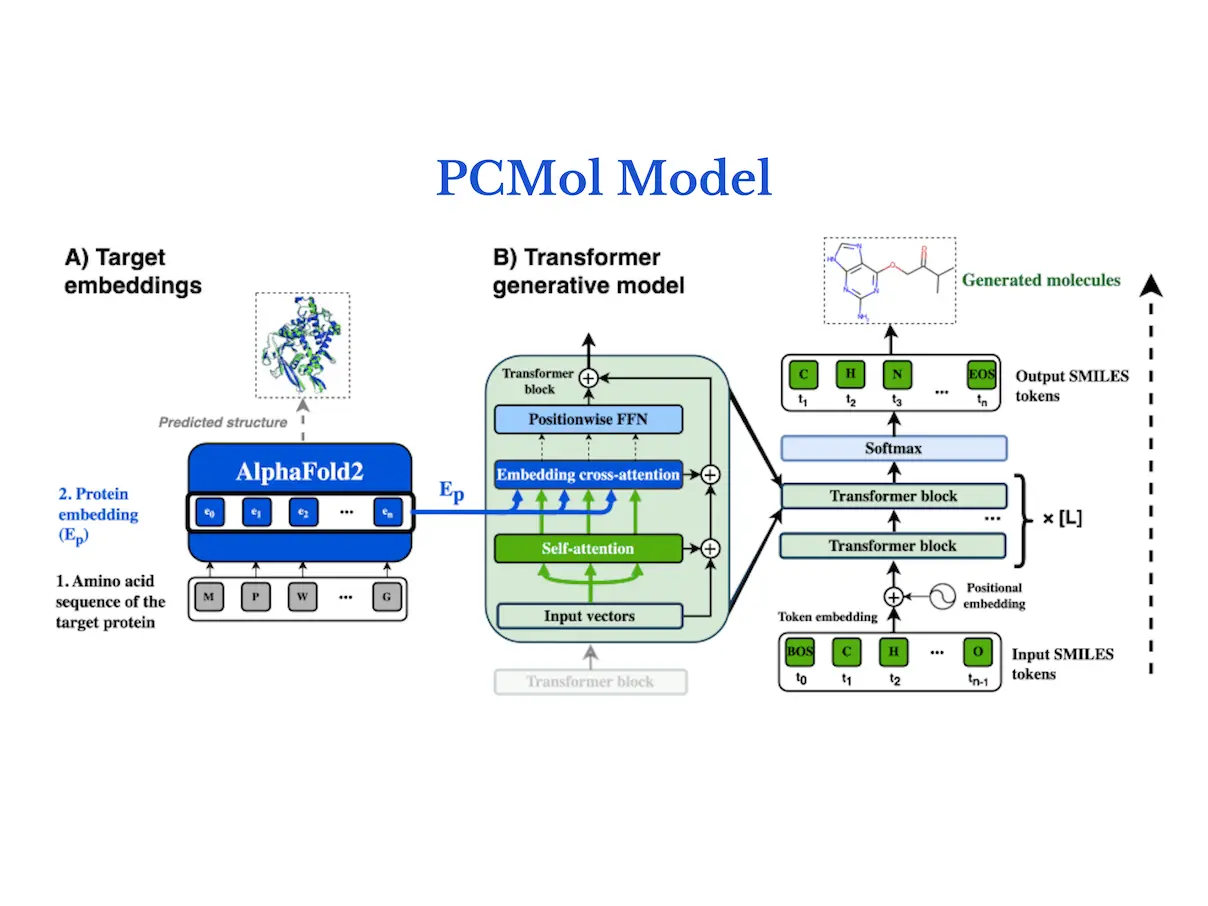
Understanding PCMol
A de novo generative transformer model is conditioned on target protein structures using a generative model called PCMol, which expands on the utilization of latent protein representations of AlphaFold. The results of the study show that PCMol can produce a variety of potentially active compounds for a broad range of proteins, including those for which ligand bioactivity data are scant. In order to demonstrate the viability of employing AlphaFold protein representations to direct the molecule generation process and improve the generalization capabilities to unknown targets, researchers additionally benchmark against current target-conditioned transformer models. Furthermore, researchers show how crucial data augmentation is for enhancing generative models’ performance in low-data regimes.
Integrating target information from protein embeddings, the model of PCMol is a transformer encoder-decoder with a cross-attention mechanism. The next token of a molecule in SMILES string format is predicted using the conventional autoregressive goal. Transformer blocks follow a Pre-LN layout, where dropout layers come before position-wise feedforward layers, and layer normalization comes before attention layers. Within the trainable parameters of the model, transformer positional embeddings are learned.
Findings of the Study
Using the PCMol approach, compounds with increased Tanimoto similarity values to known (held-out ligands) targets, like the D(4) dopamine receptor and Prelamin-A/C, are produced. But in certain situations, like with the D(4) dopamine receptor and Prelamin-A/C, the generalization performance differs. Larger databases of recognized bioactive substances contribute to this in part because they enhance chemical diversity and the likelihood of a related ligand. The PCMol model is helpful for targets with little bioactivity data because it transforms an extrapolation difficulty into an interpolation challenge. Additionally, GPCRs that have been in that target category for a relatively short period of time regularly produce compounds that are closer to unseen data.
Looking into Performance of PCMol in Training Set
The capacity of target-conditioned models to cover a wide range of chemical space and produce highly specific molecules based on the target indicates that they are effective at producing acceptable compounds for a variety of goals. Target-conditioned models may occasionally produce molecules that are the most likely statistical average of all molecules observed during training, though, as evidenced by the small percentage of created molecules that are centered around the centroid of the entire chemical space.
Four therapeutically significant protein targets were chosen for the purpose of discovering new possible ligands in order to illustrate the PCMol model’s potential utility. Based on known ChEMBL ligands, individual random forest QSAR models were trained, and 100 ligands were produced for each target using PCMol. According to the QSAR models, the total median potency of the created molecules is higher than the median of the known compounds for those targets. This demonstrates how target-conditioned models can be used to create compounds and improve the performance of a variety of targets.
Future Work
The AlphaFold protein representations themselves contain extensive 3D structural information, and researchers are investigating connecting 3D molecule representations (either voxel or graph-based) with them. To be utilized in other similar projects, the AlphaFold embeddings used for this project can be downloaded from the project’s repository. Moreover, a thorough virtual and laboratory screening for targets that a) lack bioactivity data and b) are near to targets in the training set in the AlphaFold embedding space would be the next step in verifying the model’s generalization skills.
Conclusion
The better generalization to unknown targets is made possible by the unique advantages that AlphaFold protein representations provide over conventional de novo generative models. These models detect similarities between target protein structures and lists of bioactive ligands by training on a larger dataset. They also produce chemicals targeting a variety of objectives, such as uncommon or little-explored ones. The utilization of high-quality protein representations by the AlphaFold model, a data augmentation technique, and larger models and datasets are some of the aspects that are responsible for its better generalization capabilities to unknown targets. The data augmentation method is crucial to make the model function well on targets with less bioactive data. The efficiency and performance of the sample grow with scaling up the model and dataset sizes.
Article source: Reference Paper | PCMol’s open-source package, along with a dataset of AlphaFold protein embeddings, is available on GitHub
Important Note: ChemRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}