

Drugs with polypharmacology, or substances that block several proteins, offer a wide range of uses but are challenging to create. To overcome this difficulty, researchers from the University of California San Diego created POLYGON, a generative reinforcement learning-based method for polypharmacology. In order to develop novel molecular structures, POLYGON embeds chemical space and repeatedly samples it; this results in the projected ability to inhibit each of the two protein targets, as well as drug-likeness and simplicity of production. With 82.5% accuracy, POLYGON detects polypharmacology interactions in binding data for more than 100,000 drugs. When administered at doses of 1-10 μM, de novo drugs that target ten protein pairings resulted in a loss of over 50% in both protein activity and cell viability. According to docking studies, top structures bind with low free energies and comparable 3D orientations to canonical single-protein inhibitors.
Introduction
In conventional drug development, the “one disease—one target—one drug” approach has produced many effective treatments, but it is not well suited to disorders having several possible intervention points, which may influence the etiology of the disease in part. This is especially difficult for polygenic diseases that combine functional effects across several genes in intricate biological networks, such as mental disorders and cancer.
Treating illnesses like KRAS mutant non-small cell lung tumors has increased interest in using multitherapy and polypharmacology medications. Better pharmacokinetic and safety profiles, less chance of developed resistance, and easier therapy formulation make these medicines more patient-friendly. Even though polypharmacology is still in its infancy, it has already demonstrated promise in treating conditions such as KRAS, proving its usefulness in improving patient compliance and curing disease.
Facing Barriers
The difficulty of creating a single agent that can effectively block many proteins at once has been a significant obstacle to the development of polypharmacology agents. Before appropriate hit scaffolds could be found, effective polypharmacology designs—such as those that target RET and VEGFR2 in thyroid cancer—required a significant investment of time and money. These factors have led to the majority of these compounds’ discoveries being accidental rather than planned.
Promising Future
Numerous new developments in machine learning are starting to show promise in related fields, such as the identification of medicines that already have the potential for polypharmacology (dual targeting), the systematic prediction of compound-target interactions, and the de novo synthesis of single-target inhibitors.
Understanding POLYGON
An AI-based deep machine learning model known as POLYpharmacology Generative Optimisation Network (POLYGON) is intended to generate new polypharmacology molecules programmatically. After being trained and benchmarked, this model produces de novo molecules that target cancer proteins that are synthetically lethal. Next, the model is used for the synthesis of drugs for the dual inhibition of mTOR and MEK1, which are confirmed in lung tumor cells and cell-free assays. POLYGON is a deep neural network that, like de novo human portrait production, optimizes chemical attributes in human portraits. It processes chemical formulae for molecules into a “chemical embedding” using a variational autoencoder (VAE). Each data point in this low-dimensional representation of complex inputs is given a coordinate, guaranteeing that similar inputs are near one another in the embedded space. A specific machine learning aim is accomplished by coupling the chemical encoder to a decoder, which transforms any place in the embedding back into a valid molecular formula.
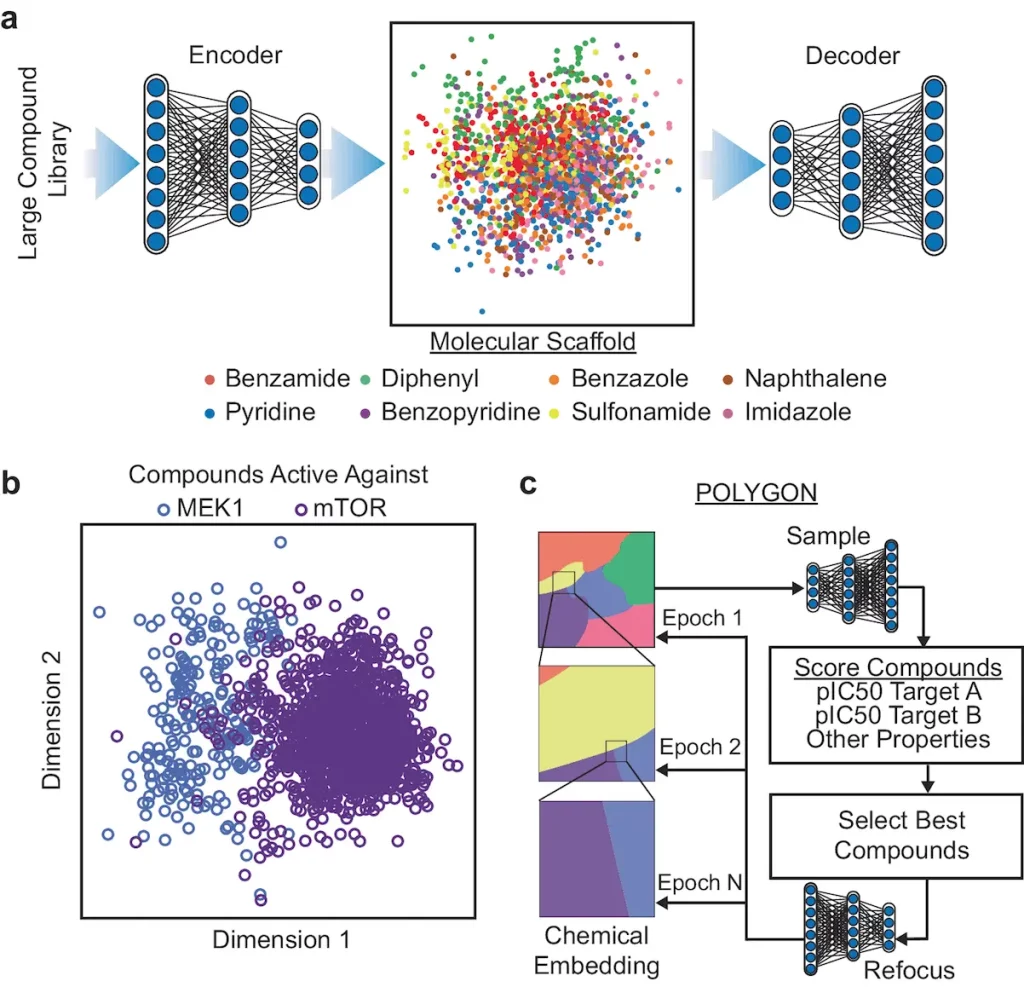
Compounds Targeting Synthetic-lethal Cancer Proteins
Using human cancer cell lines, POLYGON was utilized to create polypharmacology compounds against ten pairs of artificially deadly protein targets. Histone modifiers, tyrosine kinases, DNA binding factors, and serine/threonine kinases are some of these targets. Researchers chose the top 100 compounds for every target pair. The molecular docking studies implemented UCSF Chimera and AutoDock Vina to predict the interactions between drugs and targets. The favorable mean ΔG shift supported the binding predictions of POLYGON.
Using co-crystal structures of these proteins with their standard single-target inhibitors from the Protein Data Bank, POLYGON compounds were investigated to target the synthetic-lethal combination of MEK1 and mTOR. The mTOR-FRB/FKBP12 complex with rapamycin and the structure of MEK1 with trametinib were found. Trametinib was properly oriented within MEK1 using AutoDock Vina, with a favorable ΔG of –9.2 kcal/mol; its optimum placement within mTOR, the second target, was significantly less favorable, with ΔG of –7.4 kcal/mol. In a similar vein, rapamycin had a favorable ΔG of –8.6 kcal/mol and was appropriately positioned in the mTOR complex. It was discovered that the top POLYGON molecule (IDK12008) docked at locations comparable to rapamycin in the mTOR complex and trametinib in MEK1. Similar-looking docking results were achieved qualitatively. Qualitatively similar docking results were obtained for POLYGON-generated compounds against other synthetic-lethal protein pairs.
Conclusion
POLYGON is a polypharmacology design pipeline that generates, synthesizes, and validates compounds having activity against two targets through rigorous experimentation. In order to facilitate additional optimization using traditional methods such as structure-activity relationships (SAR), it tackles the preliminary stages of polypharmacology design. Nevertheless, at this time, POLYGON does not offer compounds that are optimized for absorption, distribution, metabolism, excretion, and toxicity (ADMET). More studies may employ SAR data gathered from synthetic drugs for successive training cycles to increase potency and selectivity against one or both targets. Incorporating IC50 estimations for off-target proteins and direct structural information of both intended and unexpected protein targets in the scoring module should enhance the generating capability of the POLYGON algorithm for dual protein inhibitors.
Article Source: Reference Paper | The source code of POLYGON is available publicly on GitHub and Zenodo.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}