
The languages of biology and chemistry are difficult for language processing and computer vision systems to grasp, notwithstanding their advances in language processing. Although LLMs can improve few-shot learning, they are not very good at capturing the connection between pharmacochemical characteristics and molecular structure, which makes small-molecule drug modification less effective in few-shot learning. Researchers from the Sichuan University, China, presented DrugLLM, an LLM specifically designed for drug design, in this paper. Group-based molecular representation (GMR) was used to represent molecules throughout the training process to enhance particular molecular features. Anticipating the next molecule based on previous modifications, DrugLLM learns to modify molecules in drug discovery. Numerous computer tests demonstrate that DrugLLM can produce novel compounds with predicted features from a small sample size.
Introduction
The capacity of small molecules to bind to certain biological targets and modify their functions makes them essential for medication discovery. Over the last ten years, 76% of all medications approved for sale by the U.S. Food and Drug Administration (FDA) have been small molecule products. The high bioavailability and ease of synthesis of these compounds, which increase their chances of reaching their intended targets in vivo, make them useful in drug discovery. However, with today’s research technologies, creating a molecule with the perfect combination of features is difficult and time-consuming. Finding a drug that works requires billions of dollars and nine to twelve years of research and development.
In order to find compounds that interact with biological targets, scientists must search over a large space, which poses a considerable obstacle to drug development. Larger experiments would be too costly; thus, computational methods are needed to focus the search. Creating completely unique and original molecules through de novo drug design is an alternative to scouring the vast space of chemicals. While deep learning and reinforcement learning techniques show tremendous possibilities in de novo drug design, traditional methods rely on laws of molecular building. Few-shot molecule production is still not well understood as the majority of existing methods need thousands of data to be learned. The development of de novo drug design methods depends on the capacity for a few-shot generation.
Understanding DrugLLM
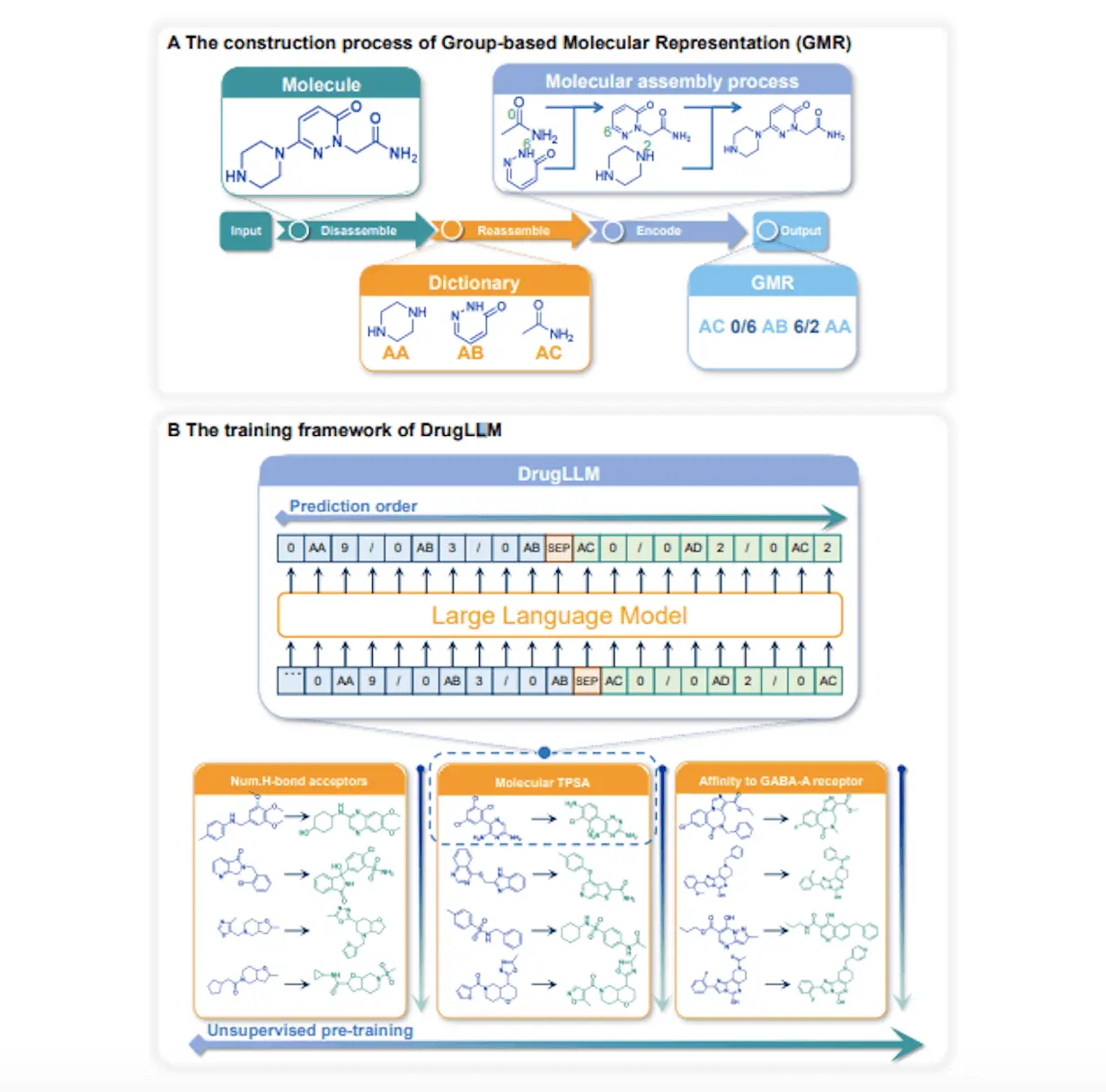
DrugLLM is a large language model for drug design that addresses token abundance, cyclic complexity, and structural sensitivity inherent in SMILES by representing molecules using Group-based Molecular Representation (GMR). GMR organizes modification sequences according to particular molecular features and converts molecules into linear sequences. DrugLLM employs a “sentence” approach in which a single case towards a certain property is considered a “sentence,” and several examples towards the same property combine to form a paragraph. DrugLLM learns the intrinsic relationship between molecular structure and related properties and uses this knowledge to continually anticipate the next molecule based on modification history.
DrugLLM: A Few-shot Learner in Molecule Optimization Towards Biological Activities
DrugLLM, a few-shot synthesis method, has demonstrated remarkable physicochemical characteristics; however, it has trouble anticipating molecules’ biological functions. Typically, these compounds are unique and not listed in the ChEMBL database. A message-passing neural network is utilized to anticipate biological activity in order to handle this. Prior to constructing the DrugLLM dataset via the ChEMBL database, ten activities with sufficient samples and a reliable property predictor are chosen. When a predictor’s forecast has a Pearson correlation of more than 0.75, it is likely to correlate well with actual statistical evaluation.
The fact that none of the three generator baselines achieve any appreciable improvement over random generation suggests that these molecular generators are still not able to capture the modification rules underlying the small number of samples. DrugLLM performs much better than the other baselines in the majority of test properties, especially in terms of producing molecules with a 76% success rate in binding to Rho-associated protein kinase 1. These results show that, given a small number of samples for an unknown chemical feature, DrugLLM is capable of deducing the intrinsic rules of molecule changes.
Limitations of DrugLLM
One of the drawbacks of the existing drug design approach, DrugLLM, is that it only offers a few-shot learning capability due to its limited input duration of nine shots. The present model lags in zero-shot learning capabilities for arbitrary instructions and only optimizes molecules towards two known molecular attributes. This is attributed to hardware restrictions. Furthermore, the existing GMR representation lacks standardization methods and, in some cases, has trouble representing a limited number of complicated molecules. Subsequent enhancements will concentrate on standardizing techniques to lower encoding errors and optimizing for unique GMR representation scenarios.
Conclusion
This paper describes the first attempt to construct a large language model for optimization and few-shot molecule creation. A large-scale textual corpus in the form of sequences of molecule alterations is built by researchers using tabular data pertaining to biological activities and molecular characteristics. DrugLLM is taught in an autoregressive fashion to forecast the upcoming molecules based on past alterations. Researchers conducted extensive computer tests and found that DrugLLM outperformed all competing techniques (including GPT4) when it came to optimizing novel compounds on more than 20 characteristics or biological activities in the few-shot learning setting. These findings demonstrate how the suggested methodology for molecule design and optimization significantly improved efficacy, underscoring DrugLLM’s potential as a formidable computational tool for drug development.
Article Source: Reference Paper
Important Note: arXiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}