
In recent years, significant advancements in machine learning, particularly AlphaFold 2 (AF2), have revolutionized protein structure prediction. These breakthroughs have generated excitement in the field of drug discovery, as accurate protein structures are crucial for rational drug design. However, the translation of these advances into predicting ligand binding poses for drug candidates remains uncertain. This article carefully examines the utility of AF2 protein structure models in predicting the binding poses of drug-like molecules, focusing on the largest class of drug targets, G protein–coupled receptors (GPCRs).
The Importance of Protein Structures in Drug Design
Since proteins are the main targets of drug discovery research, it is crucial to understand their structures in order to develop drugs that work. Determining protein structures experimentally can be time-consuming and expensive, making computational methods like docking crucial for predicting ligand binding modes. While improved protein structure prediction methods are expected to enhance ligand binding predictions, the extent of their impact remains unclear.
Evaluating AlphaFold 2 Models for Ligand Binding Prediction
This study compares the accuracy of AF2 models to traditional homology models and experimentally determined structures in predicting ligand binding poses. By systematically analyzing GPCRs, the most prevalent class of drug targets, the researchers provide insights into the strengths and weaknesses of AF2 models.
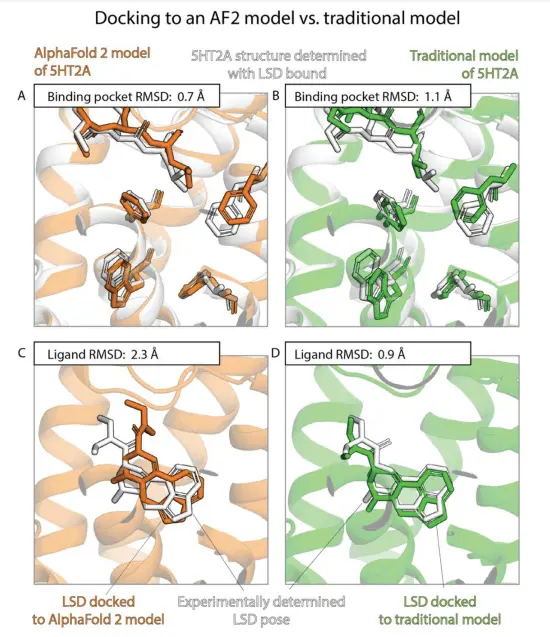
Superior Accuracy of Binding Pocket Structures in AlphaFold 2 Models
The study finds that AF2 models exhibit significantly higher accuracy in capturing binding pocket structures compared to traditional models. The discrepancies between AF2 models and experimentally determined structures are comparable to those observed between different experimental structures of the same protein with different ligands. This suggests that AF2 models excel at representing the structural details of binding pockets.
Comparable Ligand Binding Pose Prediction Accuracy
Surprisingly, the accuracy of ligand binding pose predictions using AF2 models is not significantly higher than traditional models. Docking ligands to AF2 models do not yield considerably better results compared to docking to traditional models. Moreover, ligand binding pose predictions using experimentally determined structures without the ligands bound demonstrate significantly higher accuracy. These findings indicate that AF2 models do not outperform traditional models in predicting ligand binding poses.
Implications for Drug Discovery
The study’s findings have significant ramifications for academics and business experts engaged in drug discovery. While AF2 models exhibit remarkable accuracy in capturing binding pocket structures, they do not necessarily translate into more accurate ligand binding pose predictions. Therefore, caution should be exercised when relying solely on AF2 models for ligand binding predictions. The study emphasizes the need to improve structure prediction methods to enhance their effectiveness in drug discovery.
Conclusion
Advancements in machine learning, exemplified by AF2, have significantly improved protein structure prediction. However, the accuracy of AF2 models in predicting ligand binding poses remains limited. While AF2 models capture binding pocket structures with high accuracy, their ligand binding pose predictions do not surpass those of traditional models and fall short of predictions made using experimentally determined structures. These findings provide valuable insights for researchers using predicted protein structures in drug discovery and highlight the need for further improvements in structure prediction methods.
Article Source: Reference Paper
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Learn More:



Dr. Tamanna Anwar is a Scientist and Co-founder of the Centre of Bioinformatics Research and Technology (CBIRT). She is a passionate bioinformatics scientist and a visionary entrepreneur. Dr. Tamanna has worked as a Young Scientist at Jawaharlal Nehru University, New Delhi. She has also worked as a Postdoctoral Fellow at the University of Saskatchewan, Canada. She has several scientific research publications in high-impact research journals. Her latest endeavor is the development of a platform that acts as a one-stop solution for all bioinformatics related information as well as developing a bioinformatics news portal to report cutting-edge bioinformatics breakthroughs.
- Dr. Tamanna Anwar
- Dr. Tamanna Anwar
- Dr. Tamanna Anwar
- Dr. Tamanna Anwar
- Dr. Tamanna Anwar
- Dr. Tamanna Anwar
- Dr. Tamanna Anwar
- Dr. Tamanna Anwar
- Dr. Tamanna Anwar
- Dr. Tamanna Anwar
- Dr. Tamanna Anwar
- Dr. Tamanna Anwar
- Dr. Tamanna Anwar
- Dr. Tamanna Anwar
- Dr. Tamanna Anwar
- Dr. Tamanna Anwar
- Dr. Tamanna Anwar
- Dr. Tamanna Anwar
- Dr. Tamanna Anwar
- Dr. Tamanna Anwar
- Dr. Tamanna Anwar
- Dr. Tamanna Anwar
- Dr. Tamanna Anwar
- Dr. Tamanna Anwar
- Dr. Tamanna Anwar
- Dr. Tamanna Anwar
- Dr. Tamanna Anwar
- Dr. Tamanna Anwar
- Dr. Tamanna Anwar
- Dr. Tamanna Anwar
- Dr. Tamanna Anwar
- Dr. Tamanna Anwar
- Dr. Tamanna Anwar
- Dr. Tamanna Anwar
- Dr. Tamanna Anwar
- Dr. Tamanna Anwar
- Dr. Tamanna Anwar
- Dr. Tamanna Anwar
- Dr. Tamanna Anwar
- Dr. Tamanna Anwar
- Dr. Tamanna Anwar
- Dr. Tamanna Anwar





{kind=link}