
Conventional representational models of protein functions have demonstrated notable performance in downstream tasks; nevertheless, they frequently do not incorporate explicit information on protein structure, which is essential for comprehending the connection between protein sequences and functions. To fill this void, scientists provide DeProt, a Transformer-based protein language model that can take into account the shapes and sequencing of proteins. Using millions of protein structures from natural protein clusters, the protein language model DeProt was pre-trained. It vectorizes protein structures by serializing them into residue-level local-structure sequences, which are then auto-encoded using a graph neural network. DeProt uses disentangled attention processes to integrate residue sequences with structure token sequences. DeProt works better than other cutting-edge protein language models, even with fewer parameters and training data, especially when it comes to predicting the zero-shot mutant effect across 217 deep mutational scanning assays. Additionally, it demonstrates strong representational capacities in many supervised learning tasks, indicating its broad use in protein deep learning.
Introduction
The various roles that protein sequences play in life’s complexity. Thanks to their unsupervised training on large datasets, Protein Language Models (PLMs) have transformed structural biology and bioinformatics by capturing semantic and grammatical aspects in protein sequences. Protein modeling using data-driven methodologies has advanced significantly as protein sequence databases expand at an exponential rate. PLMs have shown exceptional skill in a range of protein-related activities, proving the ability of language to represent these different functions found in protein sequences.
Although protein structures provide a deep insight into their functions, the lack of protein structure data relative to sequence data has hindered the creation of large-scale pre-trained models using structural data. Large pre-trained models based on structural data can now be developed thanks to AlphaFold2, a game-changing tool that has expanded the structural data repository to a previously unheard-of scale. ESM-IF1 and other initiatives use AlphaFold’s predicted structures to enrich datasets and improve inverse folding performance by using larger amounts of data. Direct integration of structural data into protein language models is still difficult, though, in part because overfitting can occur when there are differences between anticipated and empirical structures. Recent efforts have used quantization techniques such as foldseek to turn 3D structures into a series of discrete tokens to address this, reducing the likelihood of overfitting and making it easier to include structure data in Transformer systems. Sequence models that are built upon, such as SaProt, show that adding structural information greatly improves their representational ability.
Understanding DeProt (Disentangled Protein Sequence-Structure Model)
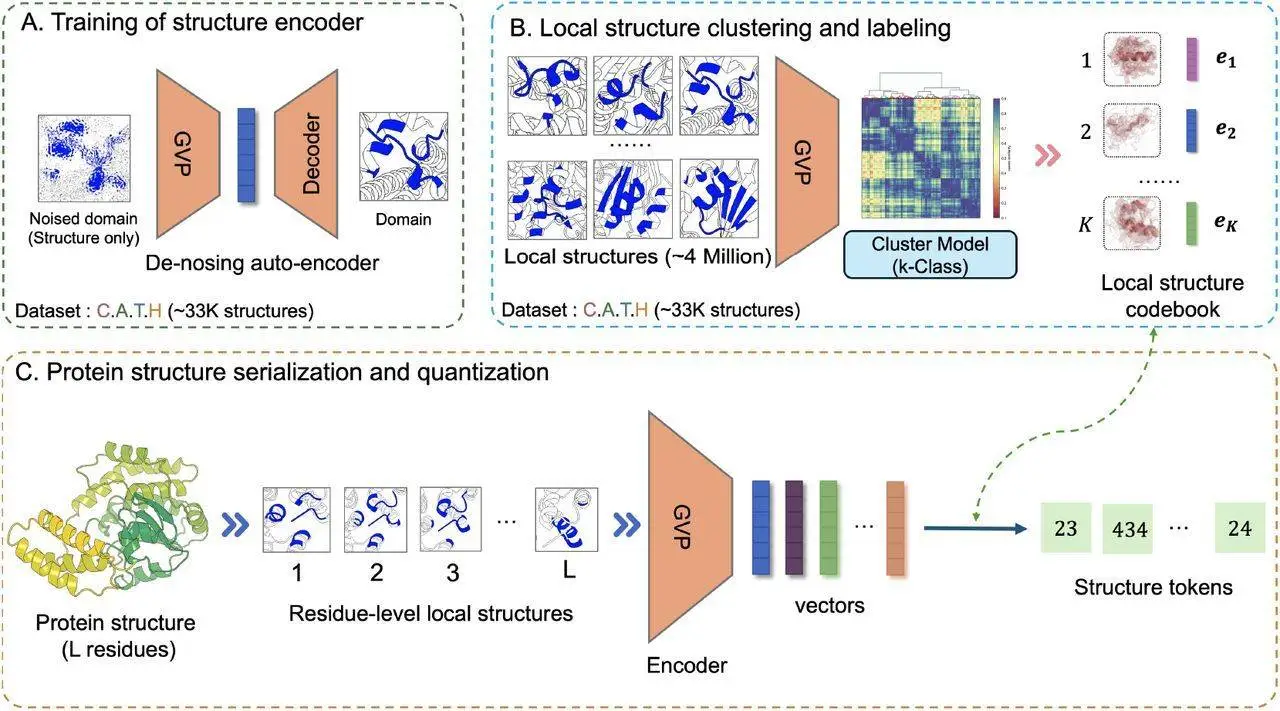
Using a Transformer architecture, DeProt is a novel framework for protein structure-sequence pre-training that aims to align protein sequences with their respective structures. This model combines sequence and structure tokens and quantized protein structures to reduce overfitting. DeProt represents a residue’s local structure using a graph neural network (GNN), which enables it to communicate with other residues in the local structure. A pre-trained codebook is used to quantize the sequences into structural token sequences after a pre-trained encoder has vectorized them. The structured token sequence and the residue sequence are fused using a novel disentangled attention mechanism, which models the ternary relationship between the primary sequence, three-dimensional structure, and relative placements of residues. DeProt demonstrates the possibility of combining structural data with protein sequences by outperforming earlier structure-aware Protein Language Models in zero-shot mutant effect prediction and downstream supervised fine-tuning tasks.
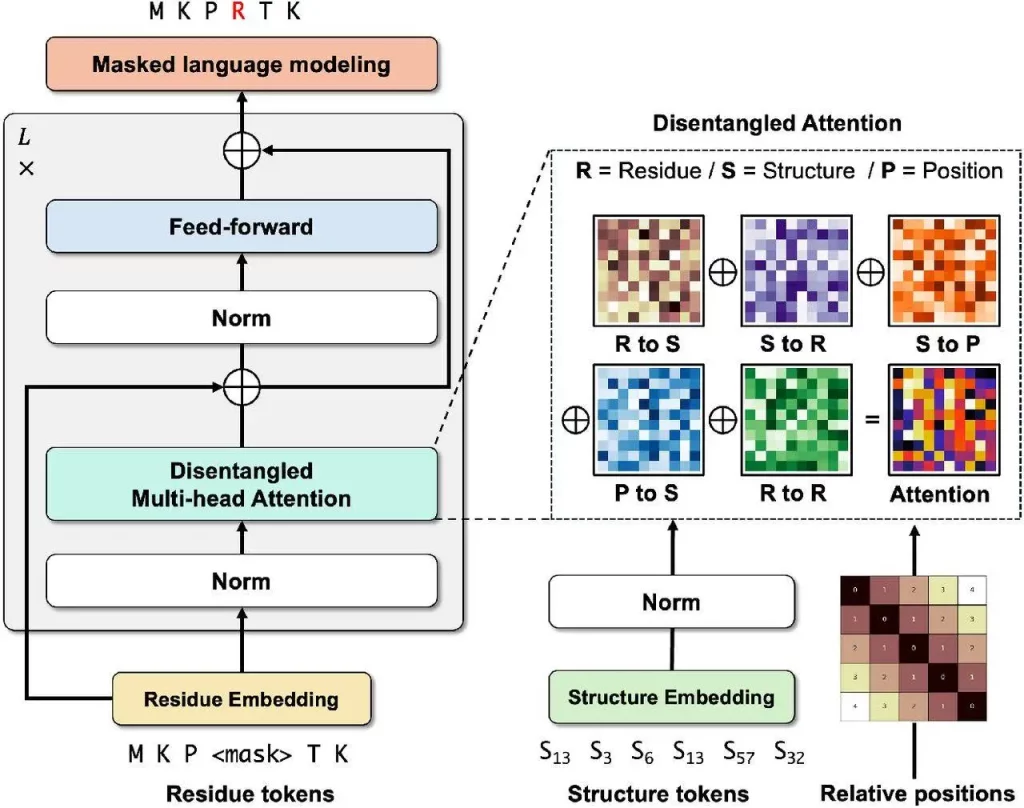
Image Source: https://doi.org/10.1101/2024.04.15.589672
DeProt’s Performance
The representation ability of the structure quantization model, DeProt, was assessed using tasks on ProteinGYM that involved zero-shot mutant effect prediction. Various function prediction challenges were used to evaluate its supervised fine-tuning skills. Key elements were identified by conducting ablation experiments to find the clustering number K, model architecture, and necessary structure information.
- In zero-shot mutant effect prediction tasks, particularly stability prediction, DeProt reaches cutting-edge performance.
- DeProt performs better in all protein-level tasks compared to previous structure-aware protein language models. This confirms that better protein representation results from including structure information in PLMs.
- The choice of K, or the structure quantization model number for clustering, affects the outcome. It turns out that K = 2048 produces the best results. In addition, the structure quantization model performs better in DeProt architecture than alternative structure serialization techniques like foldSeek and secondary structure.
- Different kinds of attention each have a unique contribution to make, but structure-to-sequence attention is very important. Making use of structure information is crucial to improving DeProt’s performance. Sequence-based protein language models remain more effective than the DeProt model unless they make use of structure information.
Zero-shot performance results
When it comes to forecasting stability in ProteinGYM’s zero-shot mutant effect predictions, the DeProt model performs better than all baselines. It works with both foldseek and DSSP, among other sequence structure quantization techniques. The structural quantization module of the DeProt model performs better than other approaches, including foldseek, which SaProt employs. Even though SaProt is built on the ESM-2 architecture, it performs competitively. The DeProt model performs best when K = 2048 is the clustering number. Performance improves as K values rise, but zero-shot performance decreases as K rises to 4096. Eliminating the structure module results in a considerable decline in the DeProt model’s performance, indicating the structure module’s critical role in the model.
Supervised fine-tuning for downstream tasks
On the majority of datasets, the DeProt model performs better than the protein language and sequence-structure models, demonstrating the significance of using structure information in protein representation models. It achieves competitive performance in supervised learning tasks and state-of-the-art (SOTA) performance in Zeroshot mutation prediction challenges by efficiently integrating structural and sequence information.
Conclusion
DeProt uses transformer-based technology and integrates both structure and residue information into its protein model. Pre-trained on eighteen million protein structures, DeProt leads the field in supervised learning tasks and reaches cutting-edge performance in zero-shot mutation detection. A de-noising auto-encoder based on graph neural networks (GNNs) is used on protein structures from the PDB data bank to introduce a unique method for protein structure serialization and quantization. A code book, which quantitatively depicts the representation vector of a protein structure, is created using the k-means algorithm. A protein structure with L residues can be quantitatively expressed to obtain structure tokens by representing it as L local structures. By avoiding over-fitting, this discrete representation technique lowers the amount of storage space required to store protein structures. The self-attention mechanism, which includes cross-attention between residues and sequence locations and residues and structure tokens, is enhanced by the disentangled attention mechanism. Despite DeProt’s remarkable performance, there is still room for development. Some suggestions include employing larger structure datasets, distillation techniques to improve extraction and encoding processes, and lowering processing costs.
Article Source: Reference Paper | DeProt code, model weights, and all associated datasets are available on GitHub.
Important Note: bioRiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}