
In this study, researchers at Icahn School of Medicine developed mEnrich-seq, a method that can enrich taxa of interest from metagenomic DNA before sequencing. This is introduced as existing technologies could only sequence the desired bacterial taxa (pathogens, beneficial microorganisms, or low-abundance taxa), and not the large background of other taxa in a microbiome sample would be useful for many applications. The fundamental principle of mEnrich-seq is to enrich target bacterial taxa while depleting host DNA and background microbial DNA through spontaneous bacterial DNA methylation. This technique guarantees that only non-digested DNA libraries are sequenced by integrating it with library preparation protocols. It has been shown that mEnrich-seq can enrich the genomic DNA of harmful or helpful bacteria, even those that are difficult to cultivate or have low abundance, from human urine and fecal samples. Its wide applicability has demonstrated that at least one commercially available restriction enzyme (RE) can target 3130 (68.03%) of the 4601 strains with mapped methylomes.
Introduction
Microbiomes are important for human health, illness, and medication response. They are excellent sources for finding genes and compounds with applications in industry, biotechnology, and medicine. However, a higher risk of pathogenic bacteria and complicated disorders is linked to the dysbiosis of microbiota or reduction of microbial diversity. Rather than sequencing every species, researchers want to investigate particular bacterial taxa in a microbiome sample. For instance, commensal bacteria are the focus of research in terms of microbe-host contact, immunological homeostasis, and biosynthetic gene clusters, whereas enteric pathogens are particularly interesting in infectious illnesses.
Microorganisms with high abundance tend to consume sequencing throughput, whereas microorganisms with low abundance are not amenable to cost-effective sequencing and research. Various technologies have been developed to meet this difficulty, including DNA binding proteins, saponin, and flow cytometry. However, these methods make it difficult to resolve bacterial species and strains with extremely similar genomes or enrich low-abundance taxa or specific taxa of interest. Our capacity to directly analyze the genomes of particular bacterial species of interest from metagenomic DNA is currently restricted as a result of these issues.
Understanding mEnrich-seq
mEnrich-seq is a method for effective enrichment of bacterial taxa of interest directly from metagenomic DNA, making use of the bacterial epigenome, or methylation of DNA, found in microbiomes. The goal of the project is to use bacterial DNA methylation in microbiomes, more precisely the bacterial epigenome, to enrich target bacterial species from metagenomic DNA. Methyltransferases (MTases), which apply methyl groups in a highly sequence-specific manner, catalyze three major kinds of DNA methylation: N6-methyladenine (6mA), N4-methylcytosine (4mC), and 5mC. Restriction modification methods are based on these motifs because they vary among species and strains within the same species. The technique uses bacterial DNA methylation motifs, enriching and reducing background DNA while differentiating bacterial species prior to sequencing.
The primary idea is to select methylation-sensitive REs individually or in combinations that logically break down the host genome and background microbial DNA at non-methylated sequence recognition sites. This removes the majority of the background metagenomic DNA while preserving the gDNA from target bacterial taxa. This method effectively directs sequencing throughput toward the analysis of microbiome-dwelling bacterial species of interest. Additionally, the study evaluated the wide-ranging applicability of mEnrich-seq, determining that 68.03% of bacterial strains, or 54.78% of the species analyzed, may be targeted with at least one commercially available RE.
Metagenomic DNA methylation-guided enrichment for target bacterial taxa
The technique of methylation-guided enrichment sequencing, or mEnrich-seq, is employed to enrich the gDNA of target bacterial species from microbiomes. In this procedure, the gDNA of the bacterial species is left intact and enriched over the background, whereas non-methylated motif sites in the majority of background DNA are cut using a methylation-sensitive restriction enzyme. The likelihood that most gDNA molecules include at least one cuttable non-methylated RE site increases with the choice of fragmentation size made during the shearing process.
Modifying the insert size and chemistry to suit various platforms allows the mEnrich-seq methodology to be used with any sequencing technology, including PacBio and Illumina. The third phase of mEnrich-seq allows for the simultaneous use of several restriction enzymes, which increases enrichment efficiency even further. Different strains or genera share certain methylation motifs, which enables targeted enrichment at various taxonomic levels.
When mEnrich-seq was used on a mock sample, the gDNA of the target bacteria was effectively enriched. The vast majority of readings that were mapped to the genome of E. coli lacked GATC sites, indicating that the gDNA molecules were able to withstand both PCR amplification and DpnII digestion. The bulk of readings containing GATC sites were found in genomic areas containing simple sequence repetitions, which are less amenable to DpnII digestion and prone to developing secondary structures. mEnrich-seq offers a novel approach to enriching bacterial taxa of interest from microbiomes despite the presence of by-product reads.
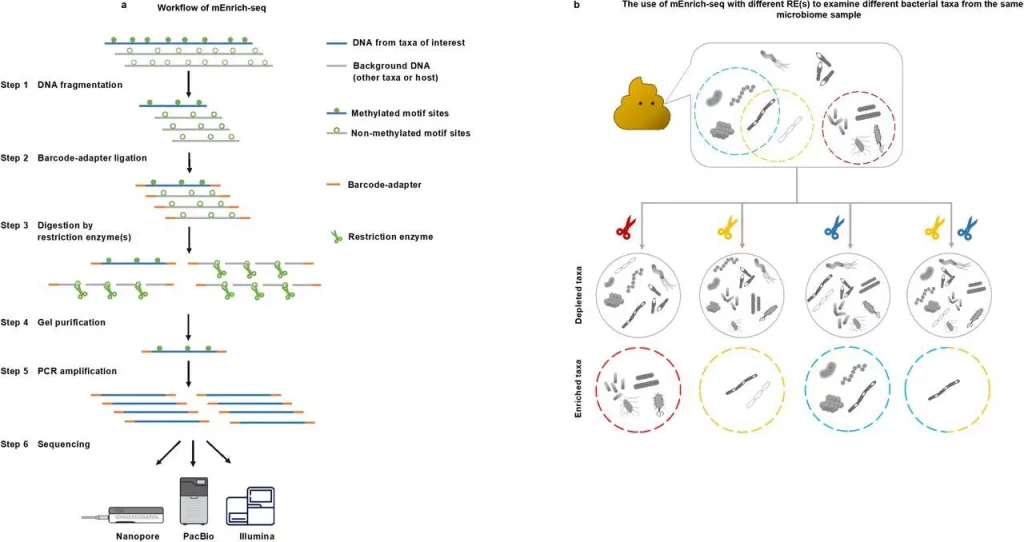
Image Source: https://doi.org/10.1101/2022.11.07.515285
Future applications for mEnrich-seq
3130 (68.03%) of the 4601 bacterial strains that were mapped as of July 18, 2022, had at least one methylation motif that may be targeted by methylation-sensitive Reactive Enzymes that are sold commercially, according to a meta-analysis of 4601 mapped strains of bacteria. These strains, which account for 54.78% of the species under study, exhibit a wide range of taxonomic variation. Since these should be easily used, the analysis concentrated on REs with recognition motifs that have 4, 5, or 6 non-degenerate bases. The application of mEnrich-seq is anticipated to extend to REs with additional target motifs with lower frequency as researchers develop more sophisticated DNA extraction techniques.
Limitations of mEnrich-seq
- Further work is required to expand the usage of commercially available REs, even if they are easily accessible to all researchers. Furthermore, not every read from mEnrich-seq comes from bacterial genomes that have the appropriate level of DNA methylation.
- Because ssDNA phages, RNA phages, and parasites are not substrates of RM systems or orphan prokaryotic methyltransferases, mEnrich-seq can only be used to enrich bacteria, archaea, and dsDNA phages.
Conclusion
The research developed mEnrich-seq, a technique that enriches target species from metagenomic DNA for selective sequencing using bacterial epigenomes. This technique enables microbiome researchers to customize their sequencing strategy to best meet their objectives by using bacterial DNA methylation to enrich specific taxa of interest prior to sequencing. It has been demonstrated that mEnrich-seq enriches potentially helpful species like A. muciniphila from fecal samples and E. coli from urine samples. It is anticipated that roughly 68% of bacterial genomes, or 54.78% of the species investigated, can be targeted by mEnrich-seq with at least one RE based on a meta-analysis of 4601 bacterial methylomes that have been mapped to date.
When combined with one or more REs, mEnrich-seq offers a customizable method for breaking down a microbiome sample. The specific combination of REs that are employed can be customized according to the taxa that are of interest in a given application. It works with various sequencing systems, such as PacBio, Illumina, Nanopore Sequencing, and others.
mEnrich-seq is a versatile and cost-effective approach for enrichment sequencing of diverse microbial taxa of interest directly from the microbiome, providing microbiome researchers with an additional approach to studying complex microbiomes more effectively.
Article Source: Reference Paper
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}