
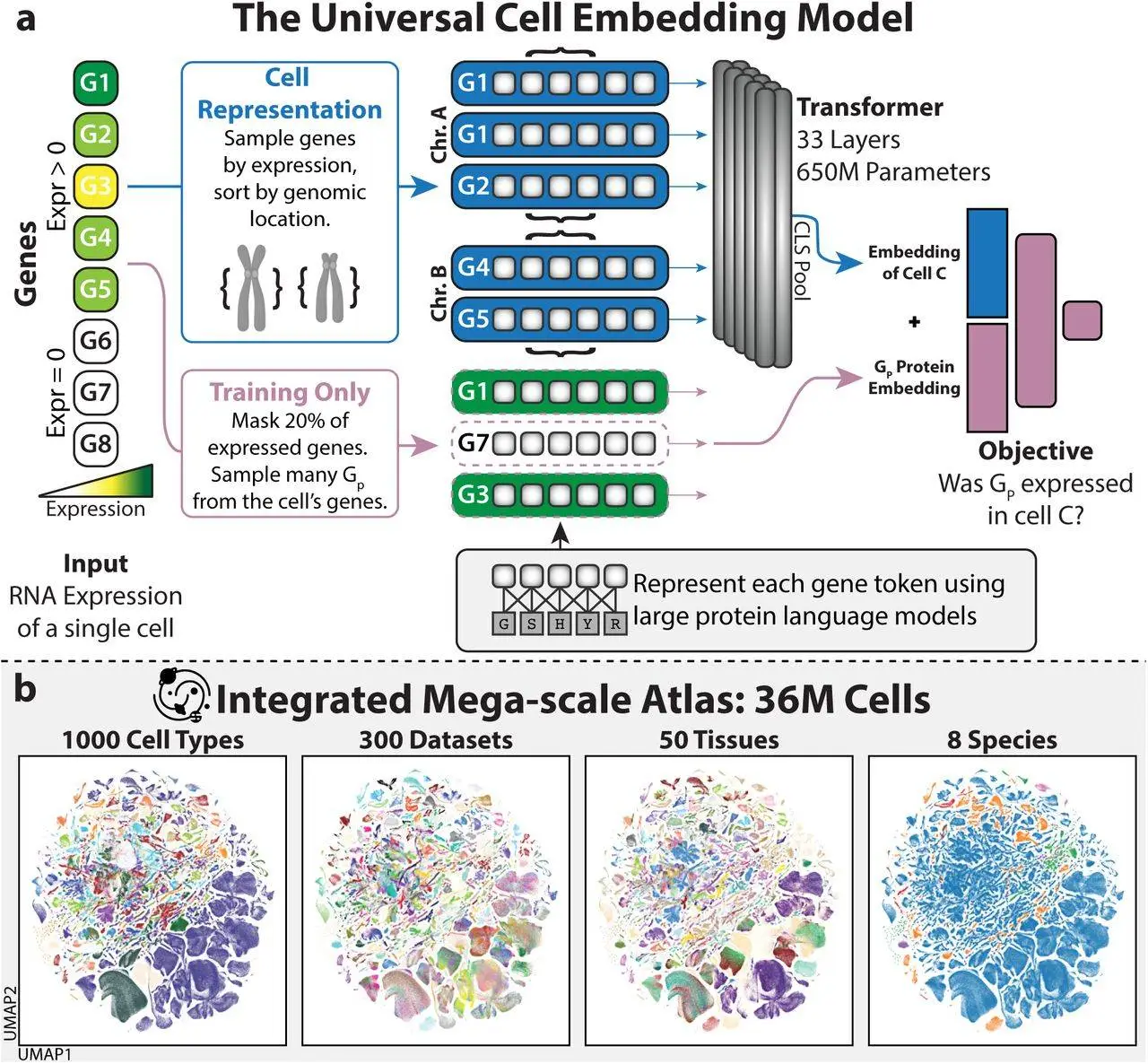
Universal Cell Embedding (UCE) is a foundation model proposed by a group of scientists from Stanford University. Using a corpus of cell atlas data from humans and other species, UCE was trained entirely self-supervised without the need for any data annotations. Regardless of tissue or species, any cell can be represented by UCE’s unified biological latent space. Despite the existence of experimental noise in a variety of datasets, this universal cell embedding effectively captures significant biological heterogeneity. One significant feature of UCE’s universality is its ability to map every fresh cell from any creature to this embedding space without the need for extra data labeling, model training, or fine-tuning.
Introduction to Universal Cell Embedding
Cells are the basic building blocks of life, and biologists have long thought of them as components of various universal landscapes; an example of one of those landscapes is known as the Waddington landscape, which illustrates the progression of cells from pluripotent phases to terminally differentiated endpoints, thus emphasizing the essential components of life and the interactions among various states in both development and illness.
The substantial growth of single-cell RNA sequencing datasets opens up fresh opportunities for computational analysis. These databases include comprehensive transcriptome snapshots across a range of tissues, donors, time intervals, and species. However, current methods find it difficult to analyze varied datasets because of limitations or artifacts peculiar to a species.
Computational methods for scRNA-seq data have overcome limitations but require model tuning for each new dataset, making representations non-universal. This requires dedicated, resource-intensive data labeling and model training for standard analyses, resulting in sub-optimal analyses based on small, limited, and private datasets, making the process time-consuming and inefficient.
Artificial intelligence has developed foundation models like ChatGPT, PaLM, Llama, and SAM, which can learn universal representations for diverse downstream tasks. These models have applications in biological contexts, such as learning protein and DNA sequences. However, their unique characteristics require specialized modeling approaches for single-cell genomics data.
Universal cell embedding was introduced to solve the above difficulties. A foundation model for single-cell gene expression, Universal Cell Embedding, is intended to answer issues in molecular and cell biology. Because UCE is resistant to batch- and dataset-specific artifacts, it can uniquely build representations of new single-cell gene expression datasets without the need for model fine-tuning or retraining. Furthermore, it accomplishes this without requiring any preprocessing of the input dataset, such as gene selection or cell type identification. Even if the genes are not homologs of those observed during training, UCE can be applied to any set of genes from any species. Through its universal description of cell biology, UCE can extend insights beyond experimentally observable facts and has intrinsic meaning. The emerging organization of cell types seen in the representations acquired by UCE is in line with accepted biological theory. With no further model retraining required, these cell embeddings demonstrate enhanced dataset integration performance as compared to current atlas-scale integration techniques, allowing for the correct prediction of cell types.
UCE provides an innovative approach for examining cell states. It allows new data to be mapped into an embedding space that is universal and already filled with reference states that have been tagged. This approach lessens reliance on small sets of marker genes to communicate findings across research and tackles problems like noisy measurements that restrict data alignment between trials. With UCE, researchers can apply pre-existing models to new data without having to retrain their models or label new data. In addition to overcoming the constraints currently encountered when dealing with small, isolated datasets, this can promote innovative cross-dataset findings. For example, a cell type classifier that has been trained to anticipate particular immune cell types can be easily applied to a brand-new dataset.
Future Horizons
New benchmarks and studies should be conducted at higher resolutions to improve UCE, and it is further noted that current models do not take raw RNA transcripts into account.
This genetic accuracy at the transcript level should be incorporated into future single-cell foundation models. These models will become more and more capable of simulating the biological functions of cells as they take on more physiologically relevant properties, eventually giving rise to “Virtual Cells.”
The UCE can be a big step forward in the development of a virtual cell. The scientists anticipate that as the amount and variety of single-cell datasets increase, UCE will prove to be a useful tool for analysis, annotation, and hypothesis development due to its ability to learn a universal representation of every cell type and state.
Illuminating Insights: The Power of Universal Cell Embedding
The application of the UCE model has led to the creation of an Integrated Mega-scale Atlas, embedding 36 million cells with more than 1,000 uniquely named cell types from hundreds of experiments, dozens of tissues, and eight species. In order to deduce the role of recently found cell types, this article uncovered new insights into the arrangement of cell types and tissues inside this universal cell embedding space. Identifying developmental lineages and embedding data from new species not in the training set are examples of emergent behavior displayed by UCE’s embedding space, which reveals new biology for which it was never specifically instructed. Overall, as the volume and variety of single-cell datasets increase, UCE offers a useful tool for analysis, annotation, and hypothesis creation by offering a universal representation for all cell states and types.
Conclusion
UCE provides a structured, diversified environment for researchers to map new data, matching cell types across tissues and species, reflecting current biological understanding. It depicts cell biology across a wide range of single-cell datasets. The UCE model has broad implications for creating large foundation models in single-cell biology. It provides a unique representation with zero-shot embeddings, thus extending insights beyond experimental data. UCE achieves generalizable representation across different datasets, comparable to methods that need retraining.
Although UCE models are constrained by coarse cell-type labeling, they allow for innovative studies of scRNA-seq data. New analyses and benchmarks need to go beyond resolution constraints in order to gain a deeper understanding of single-cell foundation models. Subsequent models ought to integrate transcript-level genetic accuracy, enabling the virtualization of biological processes and integrating fundamental biological motivation. By enabling the study of actual units of structure and function in organisms, UCE represents a substantial improvement in virtual cells and improves analysis, annotation, and hypothesis creation in single-cell datasets.
Article Source: Reference Paper | UCE source code is available on GitHub
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}