
Predicting protein structures has been transformed by artificial intelligence. However, rather than competence, accessibility, and ease of use are increasingly becoming limiting issues for end users as more sophisticated and powerful software is developed. In order to simplify the process of medium-scale protein-protein interaction prediction, the author Thomas C. McLean, from John Innes Centre, UK, presents a pipeline based on Google Colaboratory called LazyAF. This pipeline integrates the current ColabFold BATCH. Using LazyAF, he predicts the interactome of the 76 proteins encoded on a multi-drug resistance 20 plasmid with a large host range, showcasing the convenience and accessibility of the pipeline.
Introduction
AlphaFold2 and RoseTTAFold, two AI-based programs for protein structure prediction, have been incorporated by Google Colaboratory into notebook services like ColabFold BATCH. Because of this integration, big or complex protein structure predictions may now be made more quickly and with greater accessibility thanks to powerful graphics processing units (GPUs). From single protein structure predictions to screening huge datasets for novel protein-protein interactions (PPIs), the potential scope of protein structure prediction has grown. Designed to operate with ColabFold BATCH, a Google Colaboratory-based LazyAF pipeline makes it easier for individuals with less bioinformatics background to screen huge datasets for novel PPIs. The pipeline lowers the entrance barrier for medium-scale PPI prediction and enables more wet-bench researchers to incorporate in silico modeling into their workflow by performing automatic medium-throughput prediction of protein complexes in an all-versus-all scenario.
LazyAF: An Overview
A co-immunoprecipitation technique, in which a protein of interest is employed as bait to draw down its interacting protein partners from the entire cell lysate, is conceptually analogous to the AlphaFold2-Multimer-based prediction of PPIs. Before rating the chance of their interactions, LazyAF uses a protein as the “bait” and a list of additional proteins as the “candidates.” It then automatically performs an AlphaFold2-Multimer prediction between each bait and candidate. Two Google Colaboratory laptops are available for free as part of LazyAF. ColabFold BATCH, the foundation of LazyAF, is also accessible.
LazyAF Pipeline: An Overview
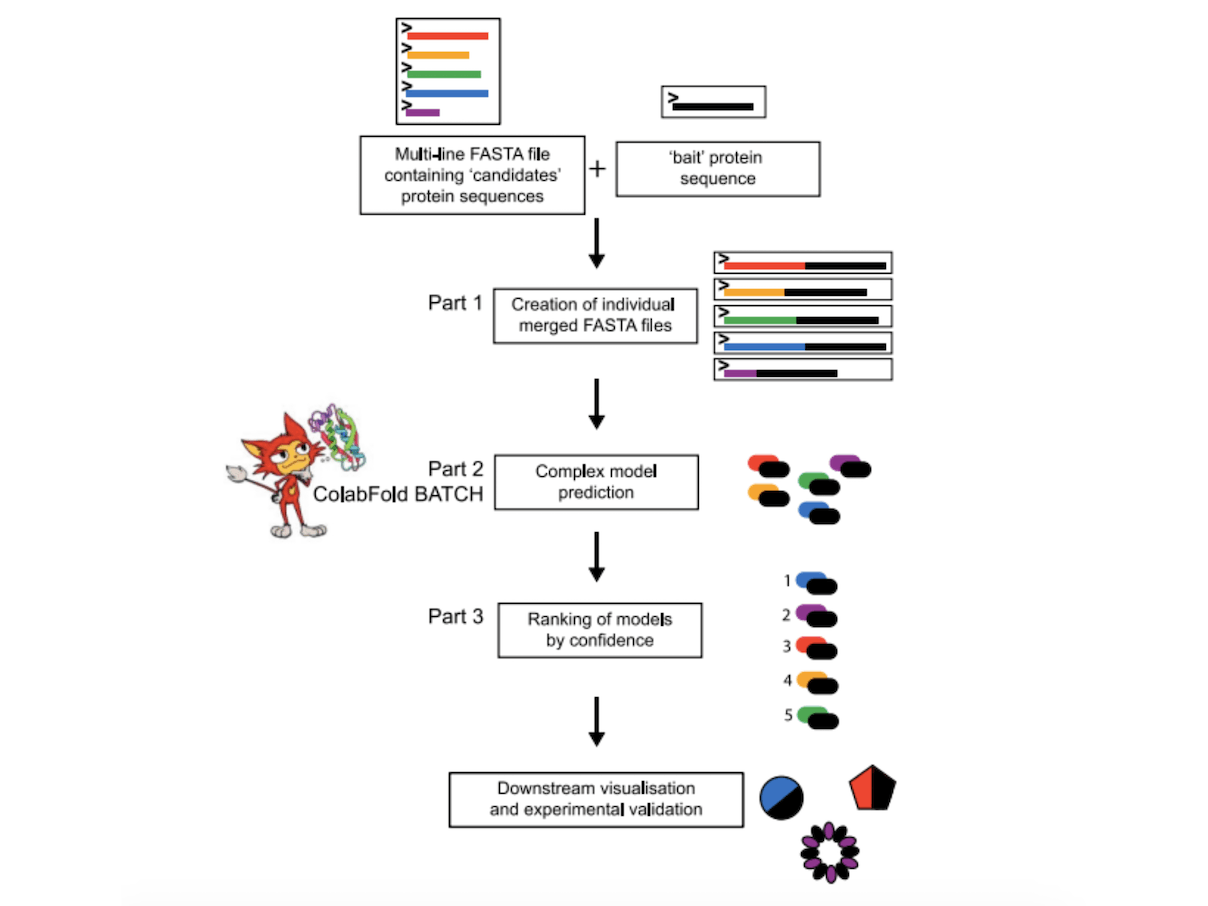
The LazyAF pipeline has three parts, Part 1 (preparation of input files for ColabFold BATCH) creates distinct FASTA files for each “candidate” protein sequence that is concatenated with a single “bait” protein sequence in a FASTA file. Then, using ColabFold BATCH, this set of distinct FASTA files is utilized as the input for AlphaFold2-Multimer-based protein structure prediction. This makes use of potent GPUs that are cloud-based, allowing for quick modeling without requiring nearby high-performance computer infrastructure. After retrieving the modeling findings from Part 2 (structure prediction in a batch mode by ColabFold BATCH), LazyAF Part 3 (ranking likelihood of bait-candidate interactions) assigns a ranking confidence score to each possibility of protein-protein interactions. Although new theories might be developed from these findings, they should always be verified through experimentation.
Data Retrieval and Processing
Each candidate protein sequence is given a unique ID by NCBI in the form of a multi-line FASTA file known as LazyAF. The name and sequence of a “bait” protein and a sequence of “candidate” proteins make up the input for LazyAF. The result is a group of distinct FASTA files, each with a unique name determined by the protein sequences of the “bait” and “candidate.” For convenience, these files are kept in a Google Drive folder on the cloud.
Machine Learning Algorithms in Action
One can utilize the LazyAF output folder as the ColabFold BATCH input directly for more details on the ColabFold BATCH approach.
Data Processing
LazyAF reduces the amount of human data processing by streamlining the analysis of ColabFold BATCH’s multiple PPI predictions. It finds and copies the JSON files linked to each co-folding’s top-ranked prediction into a new folder. From these files, the interface template modeling score (ipTM) and projected template modeling score (pTM) are taken, and for each PPI, the ranking score (ranking confidence) is computed. These rankings can be sorted from high to low for evaluating the model quality and interaction likelihood between bait-candidate protein combinations.
Prediction potential of LazyAF
Seventy-six proteins from a multi-drug resistance plasmid RK2 were used to predict protein complexes in an all-versus-all scenario using the software tool LazyAF. After being obtained from NCBI, a FASTA file containing every protein sequence found in the RK2 genome was uploaded to Google Drive. LazyAF Part 1 created 5,776 FASTA files with all conceivable pairwise combinations of protein-protein interactions using each protein sequence as a bait sequence. Google Colaboratory was used to run the notebook on High-RAM A100 or V100 GPUs. The top-ranked JSON files for each predicted PPI were copied into an analysis folder by LazyAF using the outputs from AlphaFold2 predictions as input. In addition, LazyAF computed the ranking confidence score and got the pTM and ipTM scores for every top-ranked model.
Protein A (bait) and protein B (candidate) may have different co-folding scores, according to the researcher’s observation that the heatmap of ranking confidence scores for protein candidates (PPI) is not symmetrical. This variance is anticipated as a result of the model’s iteratively refined construction. Fifty of the 5,776 predictions have a ranking confidence score in both the single and reciprocal directions that is more than 0.7. There is a strong chance associated with these anticipated PPIs. A number of these most highly anticipated PPIs, including the homodimerization of the transposase TnpA and the transcriptional regulators KorA and TetR, have been confirmed by experimentation. In order to gain a deeper understanding of the biology of the broad-host-range RK2 plasmid, the screen also revealed additional putative protein complexes that need to be confirmed and characterized by experimentation.
Step-by-step protocol to run LazyAF
- The genome of interest’s coding sequences are available at NCBI and can be downloaded in FASTA Protein format. This produces a sequence.txt file that, according to researchers, should be renamed to incorporate the name of the genome. It comprises FASTAs for every coding sequence that has been found inside the genome.
- In Google Drive, create a folder by entering its name in the My Drive area. Step 1’s sequence.txt file should be uploaded to the input folder.
- Rename files or directories, access LazyAF Part 1 in a web browser, and enter the bait protein sequence. Eliminate from the sequence any non-amino acid characters, symbols, or spaces, including a terminal *. To run the script and make sure all spaces are eliminated, select Runtime -> Run All.
Conclusion
LazyAF is a user-friendly pipeline that runs on cloud-based hardware and is compatible with any web browser. Users can manually review models, verify using complementary approaches, and evaluate factors such as per-residue confidence. LazyAF provides a simplified, user-friendly pipeline for in silico pulldown studies, in contrast to AlphaPulldown, which necessitates bioinformatic competence. LazyAF is especially helpful for wet lab scientists who wish to incorporate in silico protein structure predictions into their workflow since the distinction between traditional wet and dry laboratory research becomes increasingly clearer.
Article source: Reference Paper | LazyAF is available on GitHub
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}
[…] Revolutionizing Protein-protein Interaction Prediction with LazyAF: A Breakthrough Pipeline for Medi… […]