
Due to the rapid advancements in AI, structure-based medication design is now widely used as one of the most important methods in early drug development. AI’s deep generative modeling is expanding molecular design beyond classical methods by learning essential intra- and intermolecular interactions from available data, thanks to the increase of crystal structure data and libraries. DrugHIVE, a deep structure-based generative model, was introduced by researchers at the University of Southern California to generate a high-quality drug-like molecule. DrugHIVE enables fine-grained control over molecular generation. Molecular optimization, scaffold hopping, and linker design are high-performance approaches applied to various drug design problems using DrugHIVE.
Introduction
The early drug discovery process relies heavily on the quality of initial candidate molecules. Historically, the size of drug-like molecular libraries has been limited to a few million compounds. However, with the advent of virtually enumerated libraries and fragment-based virtual screening techniques, the capacity has increased to billions, potentially on a tera-scale. These techniques have accelerated and lowered the cost of early drug discovery but still rely on explicit enumeration of chemical libraries. Despite advancements in computational tools applying SBDD, these tasks still rely heavily on human intelligence expertise.
Generative modeling and artificial intelligence have transformed the search for chemical space, which presents a promising new strategy. De novo molecular production is made possible by deep generative models that are able to learn distributions across molecular data. Large drug-like chemical datasets have been used to apply these models, with an emphasis on drug-target interactions. These models can produce novel compounds conditioned on receptor structure by learning the joint probability distribution of ligands bound to receptors, reducing the chemical space available for drug discovery. New features like scaffold hopping, fragment growth, linker design, and molecular property optimization are added to generative models as the field develops.
DrugHIVE – Drug generating HIerarchical Variational autoEncoder
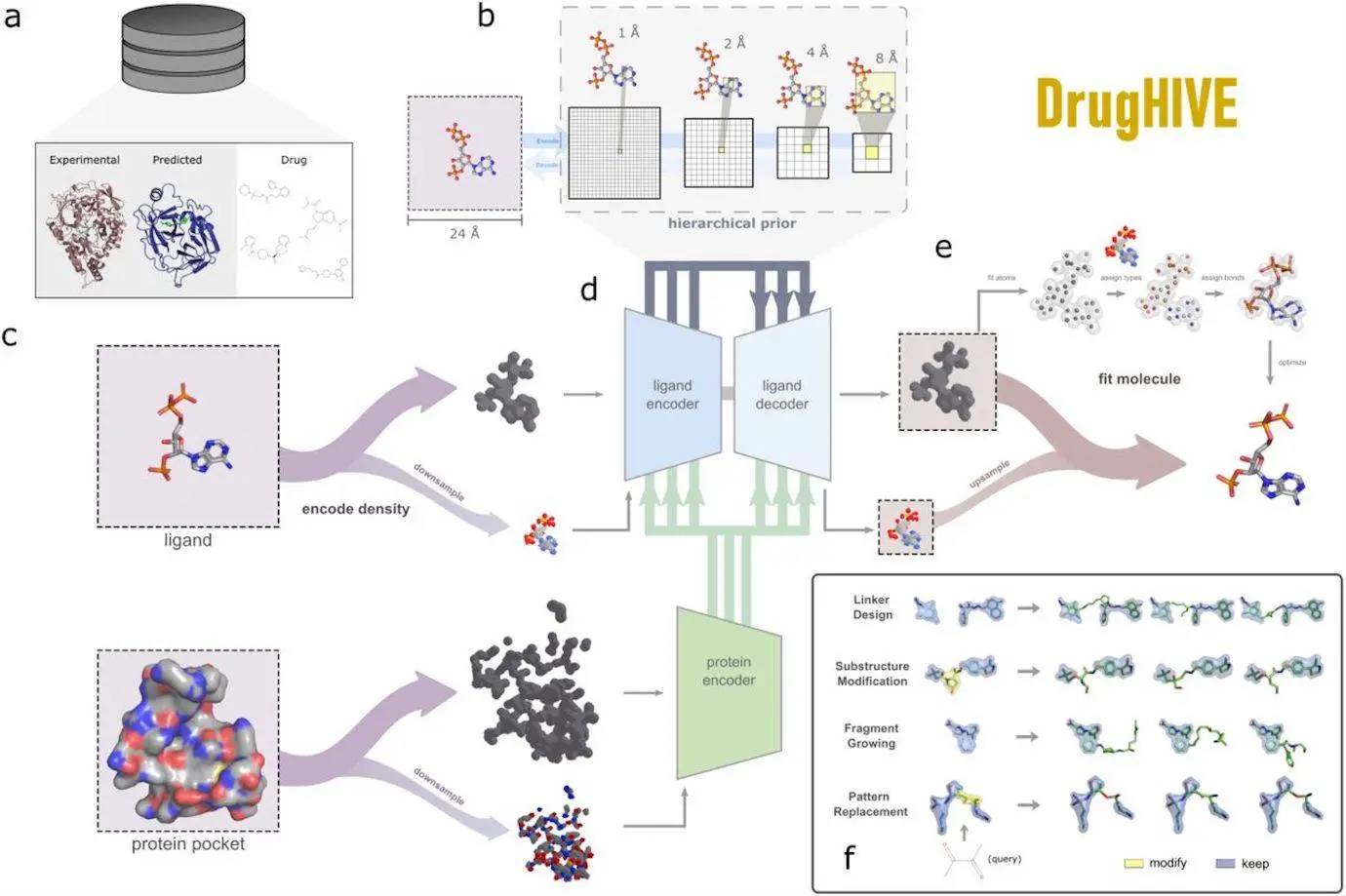
A hierarchical prior structure is used by DrugHIVE, a deep structure-based drug-generating hierarchical variational autoEncoder, to reflect the distribution of molecular structure space. This method makes it possible to create novel molecules with a high degree of spatial control, which is essential for many drug design jobs. DrugHIVE can produce molecules in a variety of ways, proving that it can handle routine drug design tasks. It has enhanced the capacity to optimize drug-like characteristics and anticipated affinity of compounds, including a novel ATM (ataxia-telangiectasia mutated) kinase inhibitor that is selective against the Vps34 kinase and the FDA-approved ACE inhibitor lisinopril. The model can also be used for fragment growing, substructure optimization, fragment linking, and automated replacement of Pan-Assay Interference Compounds (PAINS) patterns. It can also use AlphaFold2 (AF) predicted structures to produce and optimize ligands with better anticipated binding affinities for receptors, expanding the capabilities of generative molecular design beyond the collection of crystal structures that are currently available.
DrugHIVE Modelling
Molecular density grids are created from input molecules by using various atomic characteristics and multi-channel pseudo-Gaussian density to represent atoms. This work uses nine atomic elements and six additional atomic features—H-bond donor, H-bond acceptor, aromaticity, positive charge, neutral charge, and negative charge—in this work’s fifteen feature channels. Two paths are used to input the molecular grid into the network: one for atomic occupancy, which is a single-channel full-resolution grid, and another for downsampling all other atomic feature channels by a factor of two. Similar to the method used by Ragoza et al., this separation of occupancy from atomic characteristics preserves atom position information while dramatically reducing grid sparsity and memory load.
The DrugHIVE model is based on the hierarchical variational autoencoder (HVAE) architecture. The objective is to use the dataset to determine the distribution of ligand density conditioned on receptor density. However, the latent representation of HVAE inherently preserves spatial context in contrast to a typical VAE.
A molecular density grid is generated and mapped to a molecular structure using Ragoza’s atom-fitting techniques. Atom coordinates are iteratively inserted into the grid via the atom-fitting algorithm, which minimizes residual density. The density in the upsampled grid is used to assign atomic characteristics to each atom, with the highest density value across all channels being allocated the atomic number. A molecular structure is formed by the addition of bonds determined by the characteristics and geometry of the atom set.
By employing a mask to scale the reconstruction loss for zero grid values and concentrating on non-empty grid locations, the model is trained to minimize the objective.
Two datasets are used to train DrugHIVE: a subsample of the ZINC drug-like subset and PDBbind (v2020). The ZINC dataset’s subsample is utilized to enhance training and lessen overfitting, while the refined set offers excellent crystal structures of ligands attached to protein receptors. The subsample is produced by uniformly sampling 27 million molecules based on alogP and the number of heavy atoms. A dataset including 100 AF structures is utilized, and the PyMOL aligns function is utilized to analyze the data to generate the AlphaFold predicted structures. The binding pocket can be retrieved by eliminating superfluous residues and sifting out structures with low confidence scores.
DrugHIVE is a machine-learning model that creates molecular structures by utilizing various sampling strategies. The model makes use of prior sampling, in which a molecular structure is decoded using a randomly selected set of values from the prior once a protein structure has been entered. A density that fits a molecular structure is the outcome of this. Additionally, the model employs posterior sampling, in which, rather than selecting at random from the prior, the latent values from a ligand structure that has already been stored are used. Different densities of resemblance to an initial ligand density can be achieved using this method. The latent representation can be used to generate novel structures with control over molecular features because of its various scales.
Evolutionary algorithms are applied to multi-objective, non-gradient optimization issues in drug discovery. They seek complex spaces where traditional approaches are ineffective by imitating biological evolution. DrugHIVE uses prior-posterior sampling in conjunction with an evolutionary algorithm to optimize molecular characteristics. Creating an initial population of Nc molecules, applying a fitness function, and clustering them are the steps involved in the initialization stage. In the optimization step, a new population of Nc “child” molecules is created, the fitness function is applied, and the least-fit parents are swapped out for more-fit child molecules.
Before docking, the force field is optimized using RDKit; the Universal Force Field is not employed because of its poor convergence robustness. It’s crucial to avoid entering artificially stressed molecules since docking techniques, like AutoDock Vina, rely on the ligand’s intramolecular strain. Furthermore, especially with de-novo-produced structures, the inclusion of neighboring receptor atoms in computations increases the risk of convergence failure and false positives. An overly high docking score can result from omitting force field optimization.
Vina docking scores are used by AutoDock Vina, a speed-optimized variant of Vina, to estimate binding affinity for molecules. The difference between a created and reference ligand is called the Vina score, or ΔVina. Drug-likeness (QED), synthesizability (SA) scores, molecular diversity, and similarity are further measurements.
Two hundred molecules are produced for each receptor utilizing DrugHIVE preceding sampling to assess this approach. Every molecule undergoes force field optimization using the MMFF94 force field in RDKit. Plotting of the anticipated affinity against the number of atoms is done after QuickVina 2 has virtually docked to the receptor.
Exploring DrugHIVE’s functionality
- De Novo drug production using prior sampling.
DrugHIVE creates novel compounds with characteristics similar to drugs and a high projected affinity for binding to target receptors. The algorithm is evaluated against a random sample of compounds and tested on the ZINC dataset. Anticipated binding affinities of DrugHIVE-generated ligands are lower than those of ZINC compounds, with about 10.3% of them expected to have a higher affinity than the reference ligand. Twelve percent of the produced ligands have a predicted binding affinity higher than the crystal ligand for the ROCK 1 receptor, a critical target in cancer.
- Multi-objective drug property optimization using evolutionary latent search
Using prior-posterior sampling, DrugHIVE is an effective approach for producing novel ligands with great control over similarity to a reference molecule. In the encoded latent space, it enables the optimization of binding affinity, drug-likeness, synthesizability, and hydrophobicity values. This method allows for high-control, effective multi-objective optimization of molecular architectures. For instance, a considerable increase in the expected binding affinity of Lisinopril bound to the human angiotensin-converting enzyme (ACE) receptor was accomplished during optimization. Despite Lisinopril’s potent inhibition of the target receptor, the drug-likeness and synthetic accessibility scores were also raised. Latent encoding, a potent chemical space representation provided by DrugHIVE, allows for efficient multi-objective optimization of molecular structures with a high level of control.
- Utilizing Spatial Prior-posterior Interpolation for Lead Optimization
Applying prior-posterior sampling on a spatial subset of the latent representation enables molecules to be spatially transformed using DrugHIVE, a hierarchical prior structure that maintains spatial context. Common drug design issues such as fragment growth, scaffold hopping, linker design, and molecular pattern substitution are addressed with this technique. DrugHIVE can be used to connect tiny molecule fragments that have a moderate affinity for the target receptor automatically, a process known as linker design. To increase a molecule’s affinity for a target, fragment growing entails growing an existing molecule with a low binding affinity. Substructure modification, often known as scaffold hopping, is the process of improving a candidate molecule’s characteristics or expanding a candidate set by swapping out chemical scaffolds or R-groups.
Using spatial prior-posterior sampling, DrugHIVE may do automatic substructure optimization. For each fragment, it will provide a set of molecules that alter only the fragment substructure and not the entire molecule. Modifying the chemical structures that correlate to a pattern—like PAINS patterns—allows for pattern replacement, which helps prevent false negative screening decisions and false positive experimental outcomes. This feature makes drug design more effective and efficient because it can be expanded to include enormous collections of compounds.
- Drug formulation for anticipated target structures
DrugHIVE creates high-affinity compounds for receptors using solely AlphaFold predicted structures, thereby breaking a previous constraint in structure-based drug creation. The amount of protein crystal data is increasing quickly; in the last ten years, over 9,000 new structures have been added to the PDB pocket. AlphaFold2 and other sequence-to-structure prediction methods have filled in the structural gaps for foldable portions of the human proteome by providing high-accuracy structures for previously unidentified proteins. DrugHIVE ensures sufficient sequence similarity and AF confidence score by generating ligands for target receptors using both AF prediction and PDB crystal structure. Between the two sets, the range of affinities is comparable, with AF-generated ligands having a slightly greater median value. When ligands are optimized using the AF pocket by DrugHIVE, the anticipated binding affinity is higher than when the PDB pocket is used in the same optimization procedure.
Conclusion
DrugHIVE is a new approach to the structure-based design of drug-like molecules based on a deep hierarchical variational autoencoder architecture. It is based on a deep hierarchical variational autoencoder architecture. The hierarchical prior of the model enables it to produce high-affinity drug-like compounds with unparalleled spatial control and to more effectively learn the distribution of intra- and intermolecular connections. Through local exploration of the latent space, DrugHIVE can optimize the properties of molecules over a broad range of values within the context of a protein receptor.
Cycle durations can be shortened by using generative models, which automate molecular structure modification and de-novo design. DrugHIVE can be used to carry out tasks that typically call for meticulous professional attention, such as fragment growing, scaffold hopping, linker design, and high throughput pattern substitution, quickly and effectively.
DrugHIVE predicts that 10.3% of the compounds it creates for the 100 different protein structures in the test dataset will have an affinity greater than the crystal ligand. This is a substantially higher result than the 4.6% observed for a ZINC screening set that was selected at random. DrugHIVE can access previously undiscovered areas of chemical space because none of the created molecules are included in the training dataset.
Because DrugHIVE is independent of docking techniques, it provides an effective and expandable instrument for automated structure-based drug creation. Pre-clinical trial drug development could be significantly accelerated and cost-effectively decreased with further advancement and broad acceptance of these techniques.
Article Source: Reference Paper
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}