
Gene Ontology is an axiomatic theory that describes the molecular roles, biological processes, and cellular placements of proteins using more than 100,000 axioms. Learning and reasoning skills are necessary for predicting the actions of proteins in order to preserve consistency and take advantage of prior knowledge. It is still difficult to apply all of the axioms for knowledge-enhanced learning, even with the advancement of techniques. DeepGO-SE is a technique developed by researchers at King Abdullah University of Science and Technology (KAUST) that uses a pre-trained large language model to predict GO functionalities from protein sequences. Using several approximations of GO, DeepGO-SE predicts the truth values of protein functions. A neural network improves the prediction of protein function by combining truth values from several models. This strategy surpasses state-of-the-art techniques and efficiently leverages GO background knowledge to improve the precision of protein function predictions.
Key contributions of the study
- A technique known as approximation semantic entailment over multiple generated world models was created by researchers for knowledge-enhanced machine learning.
- Scientists have devised a protein function prediction technique that leverages knowledge-enhanced learning and a variety of information sources to improve the prediction performance of GO subontologies.
- Using sequence characteristics produced by a pretrained protein language model ESM2, researchers are able to identify functions for novel proteins more accurately.
Understanding Protein Function Prediction
One of the main problems in contemporary biology and bioinformatics is predicting the roles and interactions of proteins in biological systems, which makes protein function prediction an important tool. Precise functional characterizations of proteins are essential for various purposes, including therapeutic target identification, disease mechanism comprehension, and enhancing biotechnological applications in industry. Although the accuracy of protein structure predictions has increased recently, the intricacy and interactions of the few known functions make protein function prediction still difficult. The Gene Ontology (GO)2, one of the most used ontologies in biology, is used to explain the functions of proteins. Biological processes (BPO) that proteins can contribute to, cellular components (CCO) where proteins are active, and molecular functions (MFO) of a particular protein are all described by three subontologies in GO. Based on trials, researchers determine the activities of proteins and produce scientific papers, which database curators take and add to knowledge bases. Usually, homologous proteins receive these annotations. The UniProtKB/Swiss-Prot database thus includes hand-selected GO annotations for about 550,000 proteins and thousands of species.
Current approaches for predicting the function of proteins draw information from a variety of sources, including literature, coexpression, interactions, sequencing, phylogenetic analysis, and GO data. To represent amino acid sequences, the techniques can employ pretrained protein language models, sequence domain annotations, deep convolutional neural networks (CNNs), or language models like transformers and long short-term memory neural networks. Additionally, models can incorporate protein-protein interactions via graph convolutional neural networks, k-closest neighbor techniques, and knowledge graph embeddings. Additionally, automated function prediction has proved successful when natural language models are applied to scientific literature.
Limitations of Protein Function Prediction
Many function prediction techniques rely on sequence similarity to predict functions, which is one of their main drawbacks. Although this method works well for proteins that are related to one another and have well-established activities, it may not work as well for proteins that have little to no sequence similarity to functional domains that are known. Proteins with comparable structures may differ in their sequences, as molecular activities are mostly determined by structure. It’s important to remember that proteins with similar sequences can function differently based on their active sites and the species they belong to.
Methods that employ the same information sources for all three GO subontologies are therefore constrained; functions from the MFO subontology can be predicted by a protein sequence or structure, but functions from BPO and, to a lesser extent, CCO requires the presence of multiple proteins interacting in specific ways. As such, predicting BPO and CCO annotations necessitates different information sources than predicting MFO annotations. Although two proteins may have 100% sequence identity, they may participate in different biological processes based on the presence or absence of other proteins within the organism’s proteome. This is because predicting whether a protein is involved in a biological process generally requires knowledge of an organism’s proteome or at least its annotated genome. Protein-protein interaction networks have the ability to both encode the proteome and narrow down the range of possible protein interactions that result in biological processes.
Understanding the Role of Ontology In Protein Function Prediction
Protein function predictions can benefit greatly from the knowledge found in ontologies, formal theories that define a class’s meaning in a language based on logic. Knowledge-enhanced machine learning can result in better predictions from machine learning models by integrating formal assumptions from GO. Formal axioms can narrow the parameter search space by utilizing past information throughout the learning process, which enhances the precision and effectiveness of the learning process. The goal of appropriate entailment is to replicate many of the formal properties of deductive systems by explicitly performing “semantic entailment” as an optimization goal. The formal axioms of GO are only used in a small number of function prediction techniques. Subsumption axioms are used by hierarchical classification techniques such as GoStruct2, DeepGO DeePred, SPROF-GO, and TALE to extract hierarchical linkages.
Understanding DeepGO-SE
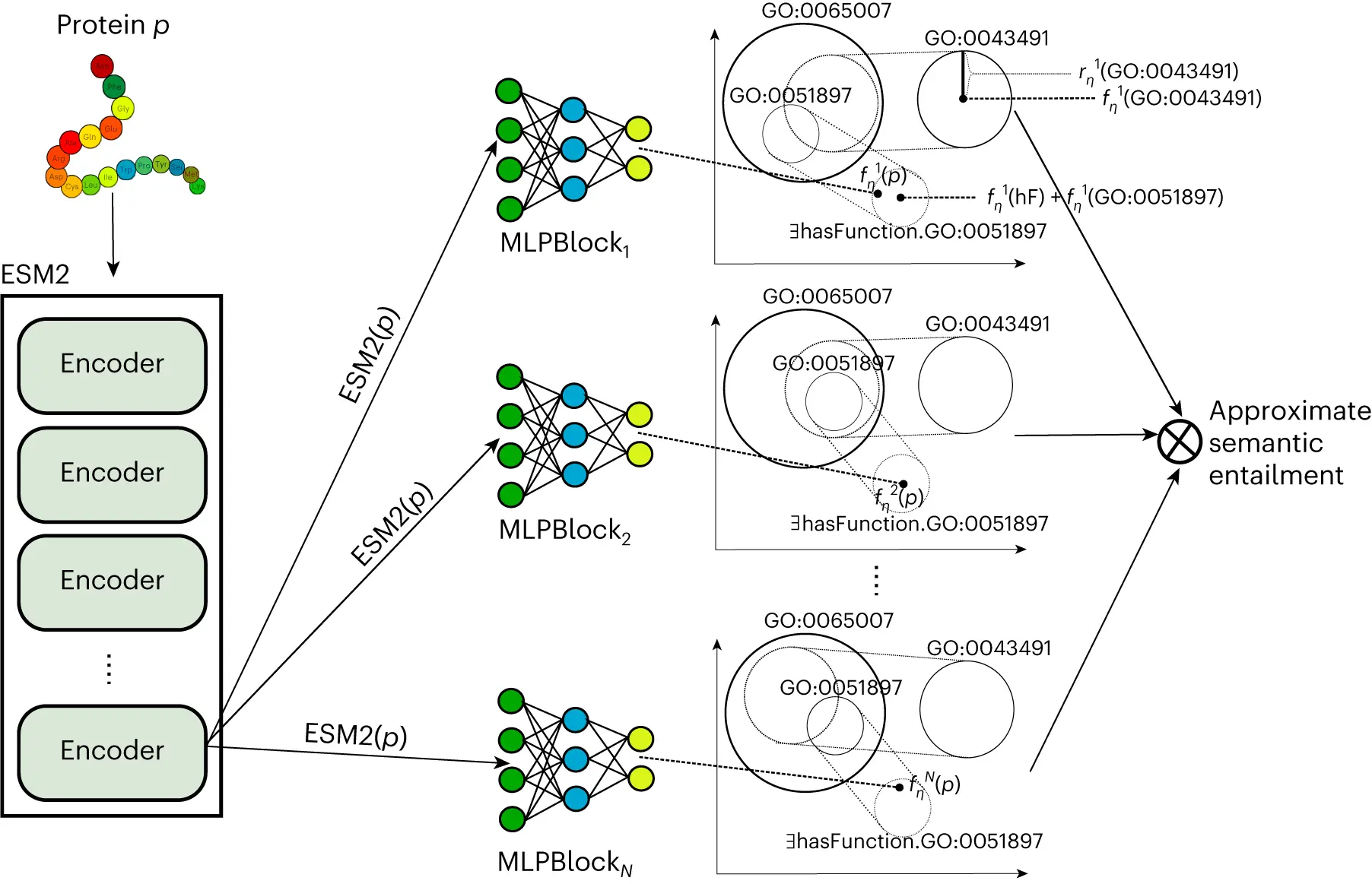
DeepGO-SE is a protein function prediction technique that uses a pretrained large protein language model in conjunction with a neuro-symbolic model that approximates semantic entailment to predict functions from protein sequences. Researchers create representations of individual proteins using the ESM2 protein language model. Researchers transfer the ESM2 embeddings into an embedding space (ELEmbeddings) that is created from the GO’s axioms, much like DeepGOZero does. ELEmbeddings correspond to a Σ algebra, or “world model,” where researchers can decide if propositions are true or incorrect. They encode ontology axioms based on geometric shapes and geometric relations.
Unlike DeepGOZero, these world models are used by academics to carry out “semantic entailment,” which states that a statement is entailed by theory T (T⊧ϕ) if and only if it is true in each and every world model where every statement in T is true. Researchers learn multiple, but finitely many, such models and generate predictions of functions as “approximate” semantic entailments, where researchers test for truth in each of the generated world models, even though there are, in general, infinitely many such world models for a theory T or a statement ϕ. Researchers demonstrate how the axioms in the extended version of GO improve the predictions of molecular activities using this kind of approximate semantic entailment.
The goal of the project is to improve predictions for intricate biological processes and cellular constituents by utilizing protein-protein interaction networks to store data about an organism’s proteome and interactome. Protein-protein interactions, as opposed to molecular activities, can greatly improve predictions of annotations to biological processes and cellular components, as demonstrated by the study. Predicting molecular interactions and functions together greatly enhances prediction performance, suggesting that molecular function knowledge alone is sufficient to predict biological process annotations rather than specific protein knowledge. Compared with sequence similarity-based techniques, the DeepGO-SE model yields much better prediction performance for all GO subontologies. Sequence similarity and a convolutional neural network (CNN) are both used by DeepGOPlus to predict functions; on this test set, DeepGOPlus’s performance suffers because it can only rely on its CNN.
Conclusion
A protein function prediction technique called DeepGO-SE improves prediction performance by taking into account relationships between proteins, background information from the GO, and characteristics of protein sequences. This methodology outperforms methods developed using a different hierarchical prediction strategy, which does not rely on prior knowledge. When paired with PPIs, DeepGO-SE performs best in predicting biological processes and cellular components based just on protein sequences. PPI predictions based on sequence and structure will be incorporated in the future. Because DeepGO-SE depends on ESM2 embeddings, it is faster than other approaches that need multiple sequence alignments and can provide zero-shot predictions. All things considered, DeepGO-SE provides a more precise, thorough, and effective method for predicting protein function.
Article source: Reference Paper | The source code of this work is freely available on GitHub
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}
[…] Unveiling DeepGO-SE: Advancing Protein Function Prediction Leveraging Language Models and GO Knowled… […]