
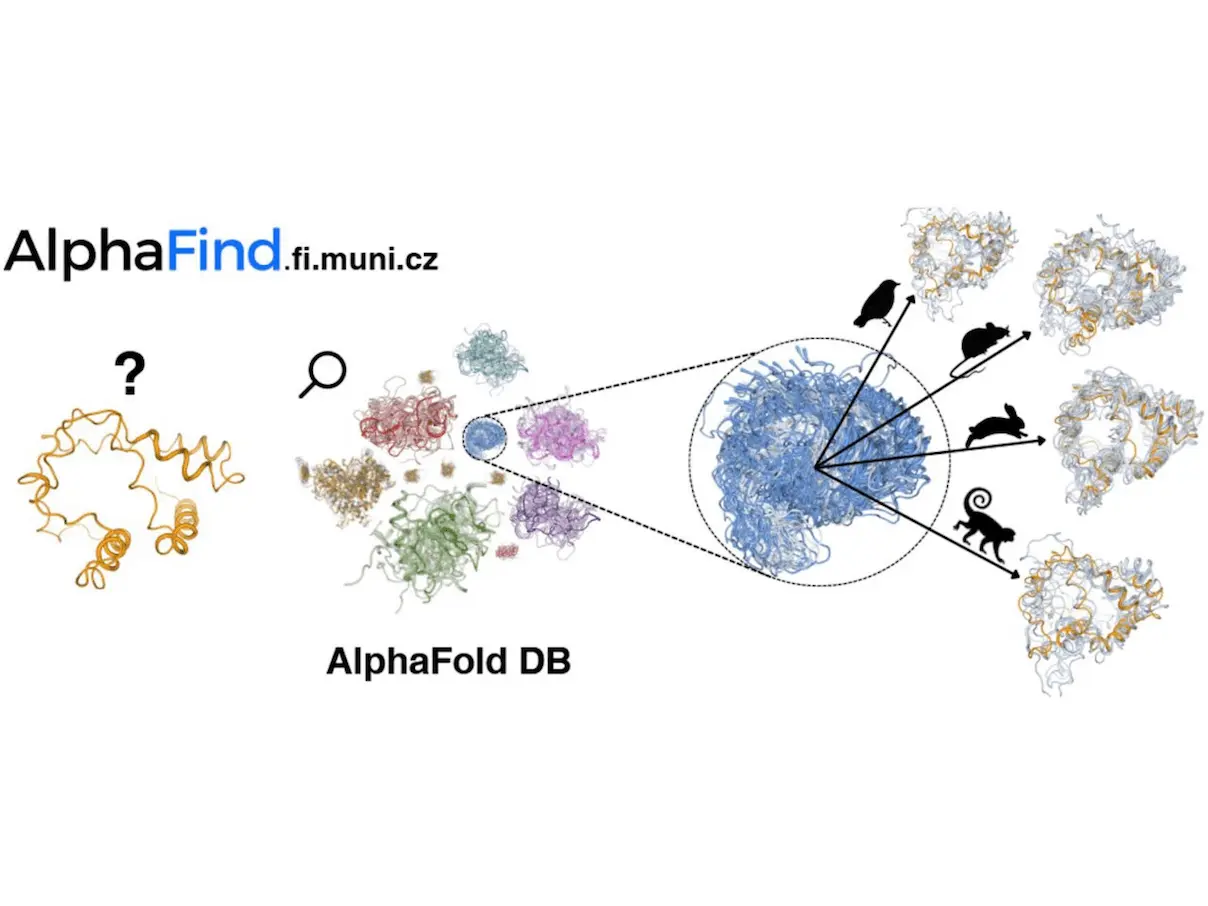
Effective structure-based search tools are necessary to efficiently organize the massive volume of data in the database. Novel tools like FoldSeek and 3D-surfer have been developed as a result of the inability of existing tools like PDBeFold to handle huge datasets. However, they are limited in their functionality by the fact that they can’t search the entire AlphaFold DB and that they have to rely on pre-established fold patterns. Because of this, researchers from Masaryk University created AlphaFind, a protein search tool that only looks at tertiary structure. It does this by automatically extracting the key 3D characteristics from each protein chain and then employing a machine-learning model to identify the structures that are the most comparable. AlphaFind is an effective protein structure tool that has shown exceptional scalability to big datasets and structures, making it possible to extract intricate and precise information.
Introduction
Protein structural data are extremely useful scientific tools that form the foundation of increasingly important and productive research. Researchers currently have about 200 thousand experimental protein 3D structures deposited in the Protein Data Bank (PDB) as a result of seven decades of intensive research. Moreover, the protein 3D structure predictions generated by the AlphaFold algorithm, which was trained using this experimental data, are extremely dependable. Consequently, the AlphaFold database, which has more than 200 million 3D protein structures, was released. Simultaneously, other databases, including 600 million predicted protein 3D structures, have been made public, such as the ESM Metagenomic Atlas. These databases are useful for many bioinformatics study areas, and new state-of-the-art technology is on the horizon.
Protein structures are massive collections of data that need to be efficiently organized and searched using their structural elements. Such massive datasets are difficult for the protein structure technologies available today, such as FoldSeek, 3D-surfer, and Dali server, to handle. AlphaFind is an application designed to search for proteins in order to address this. With an emphasis on tertiary structure, AlphaFind employs a machine learning algorithm to identify the most comparable structures by automatically extracting the key 3D properties from each protein chain. It demonstrates great scalability to big datasets and protein structures by utilizing a 3D feature extraction method specifically built for protein structures along with the Learned Metric Index approach. AlphaFind presents a viable approach to handle this massive volume of data effectively.
Understanding AlphaFind
Quick structure-based retrieval across the full collection of AlphaFold DB structures is possible using AlphaFind, an online search engine. AlphaFind is focused on tertiary structure, in contrast to other protein processing tools. It does this by automatically identifying the primary 3D properties of each protein chain and employing a machine-learning algorithm to identify the structures that are most similar. Both the AlphaFind 3D feature extraction technique and this indexing strategy have shown exceptional scalability to big datasets and protein structures. Clarity and usability were prioritized in the design of the web application itself. The searcher retrieves a list of related protein chains from AlphaFold DB, along with different similarity metrics between the query and each of the returned results. It takes as input any valid Uniprot ID, PDB ID, or gene symbol. Apart from its primary search features, the application offers researchers 3D protein structure superposition visualizations, enabling them to promptly assess the structural similarities of the items they obtain.
AlphaFind in Protein structure representation
Researchers quickly discover relevant groupings of data for a given query by capturing semantic links between protein structures using a compact data embedding method. The data analysis is more effective since this condensed representation of a protein’s 3D structure takes into consideration broad structural aspects while ignoring primary or secondary structure information.
Understanding the Workflow of AlphaFind
The useful workflow of AlphaFind has been tested for structurally similar proteins and offers an intuitive interface for efficient computational efficiency. The workflow consists of the following steps:
- Translating the input into a UniProt ID: Three types of input are supported by AlphaFind: UniProt ID, PBD ID, and Gene symbol. Other input types must be converted into UniProt ID using freely accessible APIs since UniProt ID is internally utilized to identify a protein.
- Searching for a set of candidate proteins: During data preparation, the server recognizes protein structure embeddings using UniProt ID. From 214 candidates, a fully linked neural network reduces them to about 10 million. The search space is further reduced by evaluating the Euclidean distance between each embedding and the input, which yields a list of 1000 nearest proteins.
- Evaluating global and local similarity: The closest n proteins from the candidate set are chosen based on the required result set size (n = 50, initially) in order to assess local (RMSD) and global (TM-Score) similarity, along with aligned residues and sequence identity, using the US-align tool. This step of the search process takes the longest, sometimes taking many tens of seconds.
- Downloading metadata for query and results: Once the search results are gathered, AlphaFind arranges the results by organism name for easier navigation and uses the AlphaFold DB’s API to get important data, including the name of the protein structure and the host organism.
- Visualizing protein structures’ overlap: Using NGL Viewer, AlphaFind overlays each pair of the input and resultant proteins to visually depict the similarity in protein structures. Each result also links to the Mol* Viewer for a more thorough view.
- Showing more results: The user can select to show 50, 100, 200, or 300 more potential proteins from the list of proteins found in Step 2 in order to broaden the search results. The result table is updated, and Steps 3-5 are repeated if this option is chosen.
Limitations
The foundation of AlphaFind, a tool for searching protein structures, is AlphaFold DB, a database that has been upgraded to version 3. It is capable of providing up to 1000 comparable protein structures in response to a single query; however, these are approximations and cannot contain all the most structurally related proteins. This strategy balances precision against resource requirements, which makes it appropriate for intricate data administration. Additionally, users can identify protein structures that may have gone unnoticed in earlier search results by expanding and refining the previous response with the application’s ability to load additional data. But the application treats all of the protein structure—helices, sheets, and unstructured areas included—equally, which may skew the results.
Conclusion
The program AlphaFind indexes the 214 million protein structures in the AlphaFold DB. With little back-end strain, the program quickly scans the database and returns the top 50 results. The results can be expanded by users in 15 seconds on average. Requests from users may be queued and handled slowly during peak load times; this latency may increase with concurrent user activity. AlphaFind allows users to look up similar proteins in PDB and AlphaFold DB fast. For protein structures, it employs the 3D feature extraction method and the Learned Metric Index approach. Users can download search results as CSV files, see superpositions using NGL Viewer, and sort results by TM-score, RMSD, and aligned residues. AlphaFind is platform-neutral and simple to use.
Article source: Reference Paper | AlphaFind is available online at the Website | The user manual is available on GitHub | The application’s source code is accessible under the MIT license on GitHub
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}