
Genie is a promising method for designing proteins that use expressive SE(3)-equivariant attention and simple Gaussian noise to represent structures asymmetrically during forward and backward processes. Researchers from Columbia University and Rutgers University introduced Genie 2, which is extended through architectural improvements and enormous data augmentation to capture a broader and more diversified protein structure space than Genie could. Genie 2 is a new tool for protein creation that uses a multi-motif architecture to improve motif scaffolding. This makes it possible to create intricate protein architectures with several interaction partners and functionalities. In terms of designability, diversity, and innovation, Genie 2 performs better than all existing techniques and resolves more motif scaffolding issues. These developments raised the bar for protein design based on structure.
Protein and AI
The field of protein design has grown to be important for industrial and therapeutic uses. Protein design has undergone a revolution thanks to generative AI, with models such as EvoDiff emphasizing sequence-based techniques. Amino acid polymers arranged in one dimension, called proteins, can fold into three dimensions. Discrete diffusion model EvoDiff provides an alternative to structure-based design with order-agnostic autoregressive diffusion and a denoising architecture akin to that of ByteNet.
Structure-Based Method in Protein Designing
Structure-based methods for protein function modeling include the usage of Genie, FrameFlow, and Proteus. An SE(3)-Genie uses equivariant denoiser and diffusion on backbone atom coordinates to reason across reference frames. While FrameFlow employs flow matching and adopts the basic architecture, FrameDiff uses an architecture influenced by AlphaFold for denoising. In Chroma, an effective graph neural network is combined with a correlated diffusion process, whereas in Proteus, the expressiveness of triangle attention from AlphaFold 2 is combined with faster runtimes through the introduction of graph triangle blocks and a restriction of attention to nearby residues.
The intermingling of Structural and Sequence Methods of Protein Designing
When it comes to conditional tasks that call for pre-specified sequences or structural features, merging sequence and structural elements can greatly improve protein design. Sequence information is included as a need of a structure-based diffusion process in recent approaches like RFDiffusion and MultiFlow. By fusing a discrete sequence flow with a SE(3) structural flow, MultiFlow provides diffusion or flow matching in a joint sequence-structure space. Additional research is being done on joint encoding and diffusing in latent areas.
Consideration of Protein Function in Protein Designing
Protein design prioritizes function, which is frequently controlled by a motif or residues. Proteins with particular motif scaffolding, like antigen-binding sites or enzyme active sites, have been designed using FrameFlow and other diffusion models. However, because they depend on the knowledge of inter-motif locations and orientations, existing models are unable to design proteins with several independent motifs. Proteins frequently consist of distinct functional regions that are either linked together by a flexible linker or exist as a single global domain. Designing scaffolding with many molecular functions could facilitate the creation of novel enzymes, biosensors, and medicines that alter or improve protein-protein interactions. Castro et al., 2024 inpainted an immunogen with three different epitopes using RFjoint2, a well-established non-diffusion model; nevertheless, this method has not yet been thoroughly benchmarked.
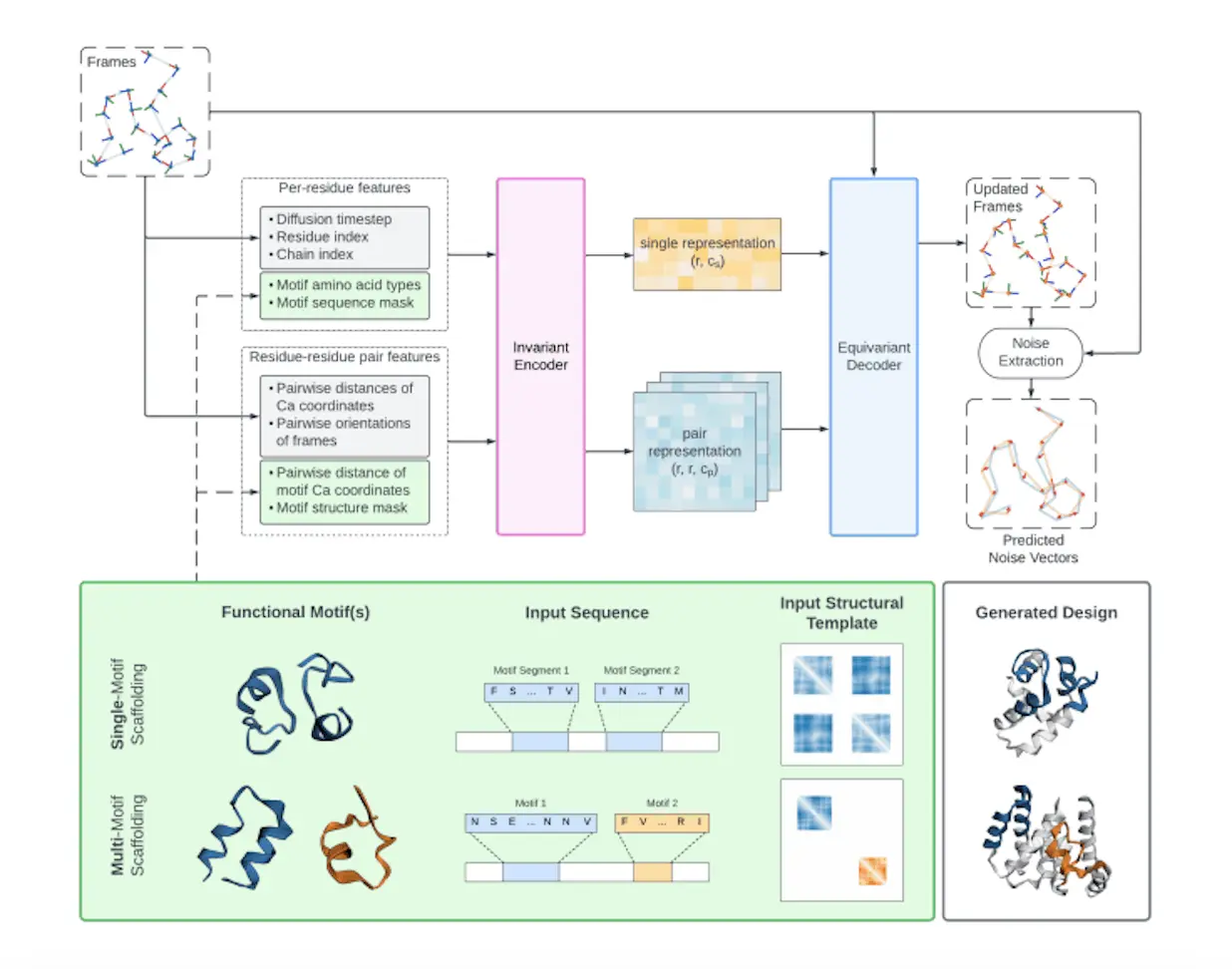
Understanding Genie 2
Researchers enable both single- and multi-motif scaffolding in this work by extending Genie. Researchers also make architectural changes and improve the training data and procedure of the main Genie model. Protein structural space is more accurately captured by the resulting Genie 2. Genie 2 achieves state-of-the-art outcomes in terms of designability, diversity, and innovation when measured against current models. Furthermore, Genie 2 outperforms RFDiffusion in motif scaffolding tasks in terms of the number of problems resolved and the variety of designs. Additionally, the researchers compile a benchmark set of six multi-motif scaffolding challenges from the literature and demonstrate that Genie 2 is capable of proposing intricate designs that incorporate many functional motifs—a challenge that protein diffusion models have not yet taken up.
Genie 2 on Unconditional Protein Generation
Chroma, FrameFlow, and RFDiffusion are two state-of-the-art models that contrast Genie 2, a protein design model. Two sets of analyses comprise the analysis: one sets the longest created protein at 256 residues without considering length, while the other sets the longest designed protein at 500 residues with length-specific limitation. Training Genie 2 on proteins with up to 256 residues length demonstrates its in-distribution generating power. The comparison is based on assessment measures and the more current model Proteus claims to have certain designability advantages over RFDiffusion at the expense of less diversity. In order to declare Genie 2 as the new state-of-the-art model, a comparison with RFDiffusion is deemed sufficient. Based on its inferior performance to ProteinMPNN, Chroma—which comes with an integrated sequence design network—is removed.
Conclusion
The most advanced technique for unconditional generation and motif scaffolding is Genie 2, yet it requires more time to sample data than previous techniques. Genie 2’s future development aims to increase motif scaffolding and unconditional protein synthesis efficiency. Introduced in AlphaFold 2, the usage of triangular multiplicative update layers has a disproportionate impact on bigger design jobs because of its high computational cost. Larger protein synthesis and training may be made possible by simplifying the Genie 2 architecture’s time and space complexity.
Article Source: Reference Paper | Genie 2 inference and training code, as well as model weights, are available freely on GitHub.
Important Note: arXiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}