
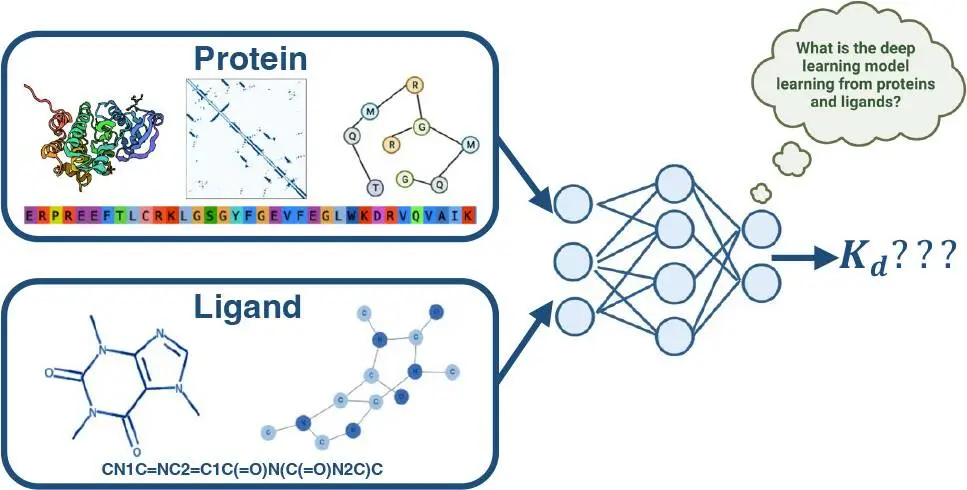
Researchers from the University of Edinburgh, UK, explore the impact of ligand and protein encodings on binding affinities prediction for different data sets, a crucial aspect of drug development and discovery. Sequence-based deep learning, convolutional neural network-based encodings, and graph neural network-based encodings were used to investigate protein roles. Graph-neural networks were used to construct ligand-based encodings. The study emphasizes the significance of incorporating ligand data into machine learning models, meaning all these techniques can enhance the reliability and speed of drug development and discovery. The findings provide key insights into how these AI systems fundamentally learn for an important drug discovery task.
Challenges and Innovations in Predicting Drug Binding Affinity
When a new drug is developed using different technologies, it is important to figure out how well a potential drug will bind to a specific protein. This helps the scientists to develop new molecules that can be used as medicine.
As it is not practical to test millions of drug-like chemicals against putative protein targets through accurate experimental screening for excellent binders, scientists have come up with different computational methods for the prediction of binding affinity over the last 40 years. This includes both ligand-based and structure-based methods, however, when screening chemical libraries against a particular protein target on a big scale, each of them still has some limitations.
Molecular docking can be used to screen vast libraries, but they frequently fall short of achieving the accuracy that a binding affinity needs. Other methods, like alchemical-free energy-based affinity prediction methods, are more accurate for finding hits in ultra-large libraries but are computationally prohibitively expensive.
Numerous initiatives have been investigated to produce ML-based solutions for binding affinity, both through the quick development of new machine learning techniques and the improved availability of binding affinity data, such as through PDBbind, KIBA, and Davis initiatives.
Deep Learning Methods for Protein-Ligand Binding Affinity Prediction
In order to understand how different components of these machine learning (ML) models affect the binding affinity prediction task’s performance, we will take a closer look at a few of these models in this study. These deep learning (DL) methods can be roughly classified as sequence-based methods or complex-based methods, depending on the kind of input data used during training.
Protein-ligand complexes in three dimensions (3D) are used to train complex-based techniques. These methods often extract information from the 3D structure of protein-ligand complexes represented as 3D Cartesian grids, interaction fingerprints, and graphs using 3D convolutional neural networks (CNNs) or spatial graph neural networks (GNNs). The binding affinity is then predicted using the properties of the retrieved protein-ligand complex.
Sequence-based methods attempt to draw insights from one-dimensional (1D) protein sequences and Simplified Molecular Input Line Entry System (SMILES) strings. Language models or graphs derived from SMILES and protein sequences can be used for this, with 2D connection information obtained from the graphs. The feature vector created by concatenating encoded protein and ligand features is used to estimate binding affinity. The 1D and 2D-based DL models extract the features from the sequence and SMILES string.
Recent Approaches in Deep Learning Architectures for Protein-Ligand Binding
Since the research focuses on the DL architectures, other examples from different research work are discussed as follows:
One of the first sequence-based techniques that used CNNs to extract 1D sequence information on the protein and ligand SMILES was DeepDTA, which was proposed by Ozturk et al. By adding more data sources, including protein domains and motifs, as well as ligand maximal common substructure terms, WideDTA expanded on DeepDTA. SMILES strings are a ligand graph’s linearized representation of its structural, geometric, and topological characteristics.
In order to depict the tertiary structure, Jiang et al. proposed a more sensible way to use the data from the 2D contact map predicted using a supervised deep learning algorithm, Pconsc4. They have also shown improvements in binding affinity performance. These contact maps, which are naturally modelable as graphs, capture the specifics of residue-residue interactions. These techniques are tested and trained on kinase data sets that are made available to the general audience. There are other sequence-based deep learning techniques that also share Jiang et al.’s design.
Current complex-based models have certain drawbacks, as Volkov et al. pointed out, including the fact that they may not always understand the physics of protein-ligand binding. They discovered that, in comparison to the corresponding interaction-agnostic models based purely on ligand or protein descriptors, the explicit description of protein-ligand interactions from complexes offers no significant advantage. Additionally, it was shown by Volkov et al. [that hidden protein and ligand biases in the PDBbind data set for complex-based model training have partially memorized the input data and have not learned the characteristics that correlate to interactions between proteins and ligands.
Experimental Design
- The ligand and protein encodings are tested in 1D and 2D.
- The two types of protein encodings are examined: one is derived from sequences, while the other is derived using contact maps.
- A one-hot encoding of the discovered binding sites is employed on the downstream binding affinity prediction task for the 1D encodings, and the ESM1-b language model’s performance is compared to that of handmade KLIFS data.
- The two-dimensional encodings based on contact maps are tested using four independent methods: protein sequence, homology information obtained from multiple sequence alignment, and AlphaFold2-predicted three-dimensional structures.
- Finally, a random contact map is employed as a control. To investigate how ligands affect the DL framework, the input SMILES string is converted into a graph structure, which is subsequently analyzed by a GNN to extract its encodings.
The impact on the downstream binding affinity prediction task is examined by examining different perturbations of the ligand graphs. The final area of research is the concatenation of the ligand and protein encodings and how this might impact binding affinity predictions. The KIBA and Davis datasets are used for all experiments.
Data Landscapes and Outcome
The researchers employed Wilcoxon’s signed rank test for error analysis and robust significance testing to ensure that statistically meaningful conclusions can be drawn when comparing our variously trained models inside the same deep learning framework. The researchers also incorporated a strong bootstrapping error analysis, which is sometimes overlooked when introducing new models. Because of this, they conducted a thorough analysis of how various deep learning model components truly affect the final downstream tasks’ performance, such as the binding affinity prediction.
This study used both 1D and 2D data to methodically examine the contributions of ligand and protein encodings to the downstream tasks of binding affinity predictions. The 1D data encodings originate from hand-curated KLIFS data for proteins utilizing DL architectures based on convolutional neural networks or from protein language models. Graph-based methods were applied to the 2D data for both ligands: SMILES strings were transformed into ligand graphs, and protein sequences were transformed into a protein contact map using either a protein structure or a protein language model.
The research stated new things about how protein and ligand embeddings routinely contribute to the learning of binding affinities for widely used data sets utilized in the literature (KIBA and Davis), even if the deep learning CNN and GCN architectures employed were not innovative.
The work effectively demonstrates that all connections learned between structure and binding affinity are through ligand encodings and that protein encodings, as they are frequently employed in the literature, make little to no contribution to downstream learning tasks.
Moreover, learning is not appreciably improved by augmenting datasets from a 1D language model to a 2D graph model, the majority of the DL techniques available today are tested and trained on small-scale kinase data sets that contain skewed binding interaction data. The choice to test DL frameworks on kinase data sets is based on the volume of available data, but careful handling of the KIBA data set is crucial to account for the uncertainty created by merging data from multiple assays and measurement types. Killiokoski’s model, which assesses the accuracy of a downstream task, lacks a single KIBA score’s reliability.
The KIBA results are useful for developing models, but caution should be exercised due to noise. Kinase data sets are a good starting point but not ideal for functional binding affinity prediction algorithms. Improved encodings for proteins can be achieved by incorporating a wider variety of proteins in data sets like BindingDB. This approach allows for the testing of novel encodings and better captures the interactions between proteins and ligands, thereby enhancing the generalizability of these models.
Conclusion
To fully comprehend protein-ligand interactions, work needs to be done in the areas of 1D and 2D encodings as well as in 3D encodings. Future research should look into jointly learned features for prediction; in this way, the DL model takes advantage of the joint features corresponding to the aspects of protein-ligand interactions. This might be accomplished, for example, by training the DL models to predict the binding affinity using physics-based 3D snapshots, much to the active learning approaches that have been investigated for adding binding affinity predictions based on molecular dynamics. The study explores the application of deep learning techniques to predict protein-ligand affinity, offering insights for medication development and discovery.
Article Source: Reference Paper
Learn More:




Deotima is a consulting scientific content writing intern at CBIRT. Currently she's pursuing Master's in Bioinformatics at Maulana Abul Kalam Azad University of Technology. As an emerging scientific writer, she is eager to apply her expertise in making intricate scientific concepts comprehensible to individuals from diverse backgrounds. Deotima harbors a particular passion for Structural Bioinformatics and Molecular Dynamics.











{kind=link}