
There’s a moment in drug discovery that every medicinal chemist knows well. You’ve found a hit, a molecule that binds to your target, shows early promise, but isn’t quite ready. It might be too big, too greasy, or just structurally awkward. So you strip it down. You keep the core that’s doing the good work, prune away the rest, and then the real challenge begins: how do you grow it back into something that actually works?
That process, called lead optimization, is where most drugs either succeed or quietly disappear. And it’s precisely where a new AI model called Sesame is trying to be useful.
The paper introducing Sesame (Spatial Evoformer for a Structure-Aware Molecular Engine) comes from Tessel Biosciences, Inc., based in Cambridge, Massachusetts. The researchers’ goal wasn’t just to build another molecule generator. It was to build one that could sit alongside a chemist, take a structural fragment as input, and grow it into a complete, pocket-fitting molecule, without needing to start from scratch every time.
The Problem With Most Generative Models
Before getting into what Sesame does, it helps to understand what most current models don’t do.
There’s been enormous progress in AI-based molecule generation over the past few years. Models can now produce chemically valid, drug-like molecules conditioned on a protein binding site. That’s genuinely impressive. But most of these systems treat molecule generation as an all-or-nothing task. You describe a pocket; the model generates a molecule. If you already have half a molecule worth keeping, there’s no clean way to tell the model: “Start here. Build from this.”
A handful of specialized tools do handle scaffold decoration or fragment linking, but they usually treat the existing scaffold as a hard constraint, a fixed set of atoms that cannot move. That rigidity creates its own problems. Real medicinal chemistry is more fluid than that. The part you’re keeping might need slight geometric adjustments. The bonds might shift. The approach needs room to breathe.
Sesame takes a different approach to this problem entirely.
Spatial Density Maps: The Key Idea
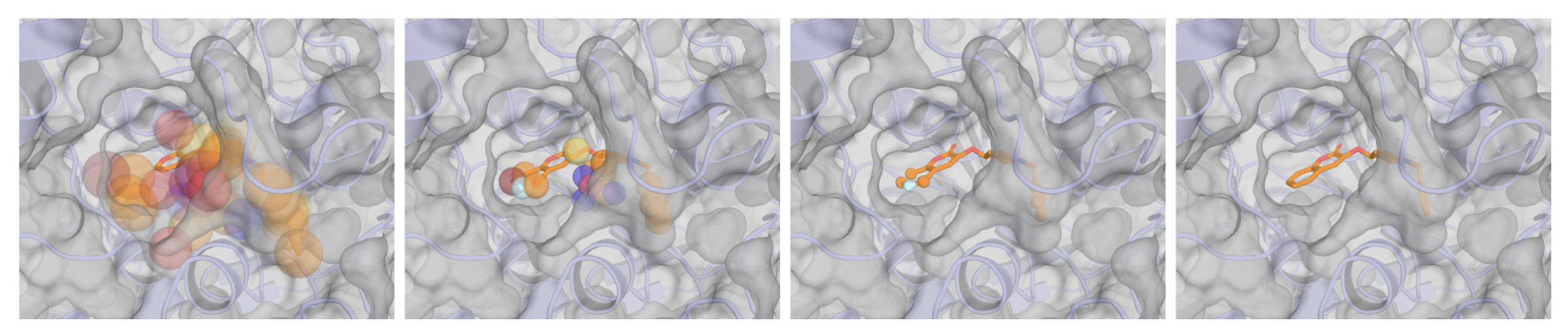
Instead of feeding a protein binding site to the model as a list of atoms and coordinates, Sesame converts both the protein pocket and any existing molecular fragment into what are called spatial density maps, three-dimensional grids that encode physical properties like charge, hydrophobicity, hydrogen bonding potential, and van der Waals forces.
Think of it as representing the molecule not as a stick figure, but as a cloud of physical influence spread across a grid. The protein pocket becomes a shaped field. The fragment you want to keep becomes another shaped field, right alongside it.
The clever part is that both inputs, the pocket and the fragment, look identical to the model. They’re just two density maps. This means the model doesn’t need a separate mode for “pocket conditioning” versus “fragment conditioning.” It handles both through the same mechanism and can handle them simultaneously. A chemist hands over a fragment as a soft prior. Sesame reads its spatial fingerprint and grows chemistry around it that fits both the fragment and the pocket.
Architecture and Diffusion
Sesame’s backbone is a MoleculePairformer, an architecture inspired by AlphaFold’s Pairformer, adapted for molecule generation. It maintains two internal representations: one for individual atoms, one for atom pairs (which encodes bonds and geometry). These representations are updated through a series of layers that blend triangle attention operations with a novel density-map conditioning module.
That conditioning module works by sampling 1,024 points from the density grid through a learned attention mechanism, then pulling features from those locations via trilinear interpolation. Those features feed back into the model’s atom and bond representations through cross-attention. Essentially, at every layer, the model is asking: “What does the space around me look like, and how should I adjust?”
The generation process itself is a diffusion model, but with a twist. Atoms have both discrete properties (type, bonding) and continuous ones (position in 3D space). Sesame handles both simultaneously within a single diffusion framework, using separate noise schedules for atom types, bond types, and coordinates, all denoised jointly. An additional finetuning step trains the model on its own generation rollouts, helping it recover from the kinds of small errors that accumulate during the reverse diffusion process.
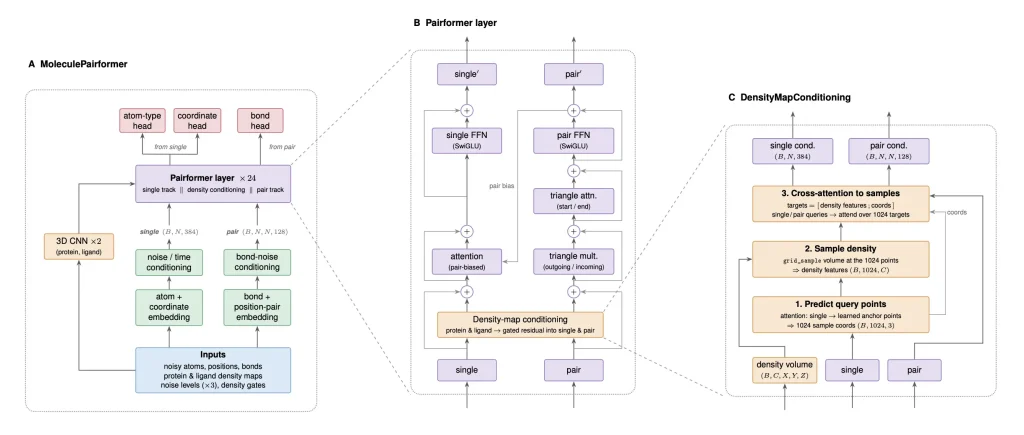
Image Source: https://doi.org/10.48550/arXiv.2606.23856
What the Results Show
The numbers are encouraging. After finetuning, Sesame achieves molecule validity rates of 92.4% for the protein-plus-fragment (lead optimization) setting and 88.7% for de novo protein-conditioned generation, after simple post-processing to remove isolated spurious atoms.
More importantly, 94.8% of fragment-conditioned molecules retain the seeding fragment as a substructure. That’s the metric that matters most for practical lead optimization: when you hand the model a scaffold, does it actually keep it? For nearly all of its outputs, Sesame does.
Property distributions of generated molecules were also compared against real drug-like ligands from a large structural dataset. Sesame tracks closely across molecular weight, lipophilicity (cLogP), polar surface area, and drug-likeness scores. Fragment-seeded generation actually performs slightly better than de novo design on these metrics, which makes intuitive sense. Starting with a known-good scaffold constrains the chemical space in a useful way.
Why This Matters
The real promise of Sesame isn’t any single metric. It’s the workflow it enables.
A medicinal chemist identifies a hit, prunes it to the pharmacophore worth keeping, and hands that fragment to Sesame. The model doesn’t demand a perfectly defined scaffold or a complete atom list. It reads the fragment’s spatial character and grows the rest of the molecule to fit, respecting both the structural seed and the pocket environment.
That’s not how most AI tools currently work in drug discovery. Most require the human to adapt to the model’s input format. Sesame is designed to work the other way around: the chemist’s insight becomes the starting point, and the model takes it from there.
Whether that translates into real-world drug discovery gains remains to be shown. But as a design philosophy, one that treats human structural intuition and generative chemistry as composable rather than competing, it’s a genuinely interesting step forward.
Article Source: Reference Paper | Web
Disclaimer:
The research discussed in this article was conducted and published by the authors of the referenced paper. CBIRT has no involvement in the research itself. This article is intended solely to raise awareness about recent developments and does not claim authorship or endorsement of the research.
Important Note: arXiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Anchal is a consulting scientific writing intern at CBIRT with a passion for bioinformatics and its miracles. She is pursuing an MTech in Bioinformatics from Delhi Technological University, Delhi. Through engaging prose, she invites readers to explore the captivating world of bioinformatics, showcasing its groundbreaking contributions to understanding the mysteries of life. Besides science, she enjoys reading and painting.











{kind=link}