
Researchers at the Abdul Jameel Clinic for Machine Learning in Health, MIT, have developed a novel method, DiffDock, an accelerated drug discovery pipeline using a diffusion generative model (DGM)-based approach for molecular docking. One of the crucial processes involved in a drug design pipeline involves molecular docking, which predicts the binding structure of a ligand to a protein. Previous methods have considered the molecular docking problem as a regression problem, and this has led to a significant increase in efficiency in terms of decreased runtime as compared to traditional search-based approaches. However, these methods are limited by their lack of accuracy. The authors developed a molecular docking method as a generative model based on the diffusion generative model approach. Given a ligand and protein structure, DiffDock learns distribution over ligand poses and has been shown to significantly outperform previous state-of-the-art docking and deep-learning methods
Previous molecular docking approaches and the paradigm shift, DiffDock
Computational drug design has aided drug discovery in leaps and bounds, and one of the crucial steps in it is molecular docking. Predicting the orientation, position, and conformation of a ligand (in this case, a drug molecule) when bound to the target protein has been dealt with in several methods. Traditional search-based approaches are based on estimating the correctness of a structure or pose using scoring functions. The algorithms then typically use an optimization technique to look for the global maximum of the scoring function. However, given the vastness of the search space coupled with the undulated landscape of the scoring function, these methods are rendered slow and often inaccurate for high throughput workflows.
Recent approaches such as EquiBind and TANKBind are deep learning-based methods treating docking as a regression problem. They predict the binding pose in one shot and are significantly faster than the search-based methods. However, they are yet to demonstrate significant accuracy improvements. According to the authors, the regression-based paradigm is not the best choice for molecular docking method development, and a generative model-based approach is more suitable. The authors, thus, develop DiffDock, a diffusion generative model-based approach that takes as input a ligand and protein structure and then learns a distribution of ligand poses.
A brief overview of DiffDock: DGM-based method for molecular docking
DiffDock is based on the diffusion generative model paradigm. Such models are capable of generating similar data as the data they are trained on. This is achieved by first adding noise to the data and then learning to recover the data by denoising. The latest sensation, DALL-E2, which is capable of generating images from natural language, is based on this paradigm.
The authors train a confidence model which generates confidence estimates for the sampled poses from the DGM and then pick out the most likely sample. The authors were thus able to find a middle path between the search-based paradigm and the one-shot prediction paradigm. DiffDock is capable of comparing and considering multiple poses as well as is not limited by the difficulties imposed by high-dimensional searches.
The model implementation involves formulating a novel diffusion process over the space of ligand poses corresponding to the degrees of freedom, viz, translational, rotational, and torsion. The set of allowed ligand pose transformations gives access to the space of ligand poses, and the DGM is efficiently trained to predict novel ligand poses.
The following image illustrates the DiffDock methodology.
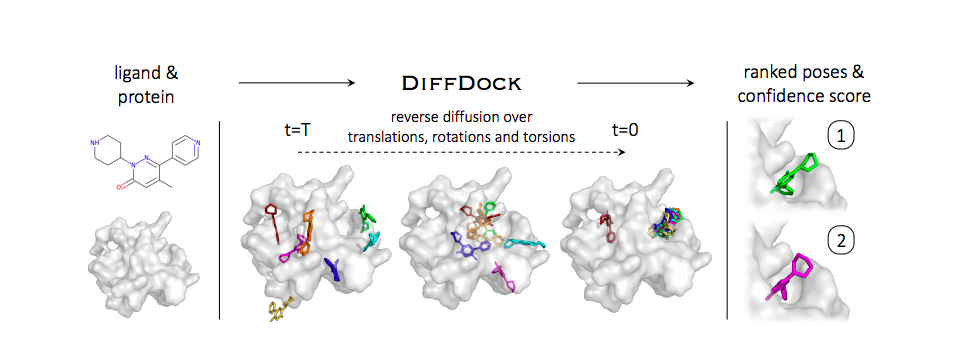
Image source: https://arxiv.org/pdf/2210.01776.pdf
Performance and accuracy of DiffDock
DiffDock has successfully achieved increased efficiency in terms of decreased runtime of the deep learning-based methods as well as significant improvement in accuracy over the search-based methods. DiffDock obtains a 38% top-1 success rate on PDBBind, better than all previous state-of-the-art methods. DiffDock maintains significantly higher precision than previous computational methods for docking. DiffDock is faster in inference time and provides confidence estimates with high selective accuracy.
Conclusion
The authors developed DiffDock based on the diffusion generative model paradigm for molecular docking and thus aiding drug design and discovery. Computational drug designing has seen significant improvements with machine learning and deep learning-based method development for the life sciences. Molecular docking greatly enables the discovery of novel drugs binding to target proteins involved in several life-threatening diseases. While experimental setups for ligand binding analysis for novel drug design are relatively slow and very expensive, computational molecular docking methods have greatly expedited the drug designing pipelines. DiffDock has been able to find the middle path between the search-based methods for docking and the one-shot prediction methods based on deep learning with the best of both worlds- the high precision in terms of predicting multiple ligand poses as well as increased efficiency in terms of greatly reduced runtime. The article was published as a conference paper at the ICLR in 2023 and promises to greatly change the drug design pipeline development owing to its efficacy and accuracy. DiffDock is a truly groundbreaking technological gift of the machine learning paradigm to the drug design community that will speed up novel drug design with reduced expenses.
Article Source: Reference Paper | Reference Article | DiffDock: GitHub
Learn More:




Banhita is a consulting scientific writing intern at CBIRT. She's a mathematician turned bioinformatician. She has gained valuable experience in this field of bioinformatics while working at esteemed institutions like KTH, Sweden, and NCBS, Bangalore. Banhita holds a Master's degree in Mathematics from the prestigious IIT Madras, as well as the University of Western Ontario in Canada. She's is deeply passionate about scientific writing, making her an invaluable asset to any research team.





{kind=link}