
For scientists studying the functions of the genome, DNA embedding is a vital tool. Effective investigations like species categorization and metagenomics binning are made possible by converting unprocessed DNA sequences into comprehensible numerical representations. Modern embedding models, however, often require refinement, particularly when dealing with unlabeled data or sequences prone to errors. Researchers introduce DNABERT-S, a genome foundation model that specializes in creating species-aware DNA embeddings. To encourage effective embeddings to error-prone long-read DNA sequences, they introduce Manifold Instance Mixup (MI-Mix), a contrastive objective that mixes the hidden representations of DNA sequences at randomly selected layers and trains the model to recognize and differentiate these mixed proportions at the output layer. Utilizing the suggested Curriculum Contrastive Learning (C2LR) technique, it may be improved even further. Empirical findings on eighteen different datasets demonstrated DNABERT-S’s outstanding capabilities. It doubles the Adjusted Rand Index (ARI) in species clustering, significantly increases the number of properly classified species in metagenomics binning, and beats the best baseline in 10-shot species classification with only a 2-shot training.
Introduction
Traditional DNA embedding models face similar challenges when attempting to comprehend a work of literature by studying individual notes without understanding the theme. The introduction of genome foundation models such as DNABERTs and HyenaDNA heralded a new era in genomic research. Large-scale pre-training has improved these models’ domain knowledge, and they have proven to be very effective in a wide range of genomic research tasks, especially when labeled data is available. However, a substantial impediment remains: there is insufficient labeled data for numerous crucial applications, including metagenomics binning. They turn sequences into numbers, but the “meaning” of those numbers, namely species information, might be lost.
This becomes particularly important when:
- Labeled data is scarce: Many genomic datasets lack substantial training labels, limiting model fine-tuning for specific applications.
- Long-read sequences are error-prone: Although new sequencing methods provide longer DNA reads, they frequently have greater error rates. Existing models fail to handle these chaotic sequences efficiently.
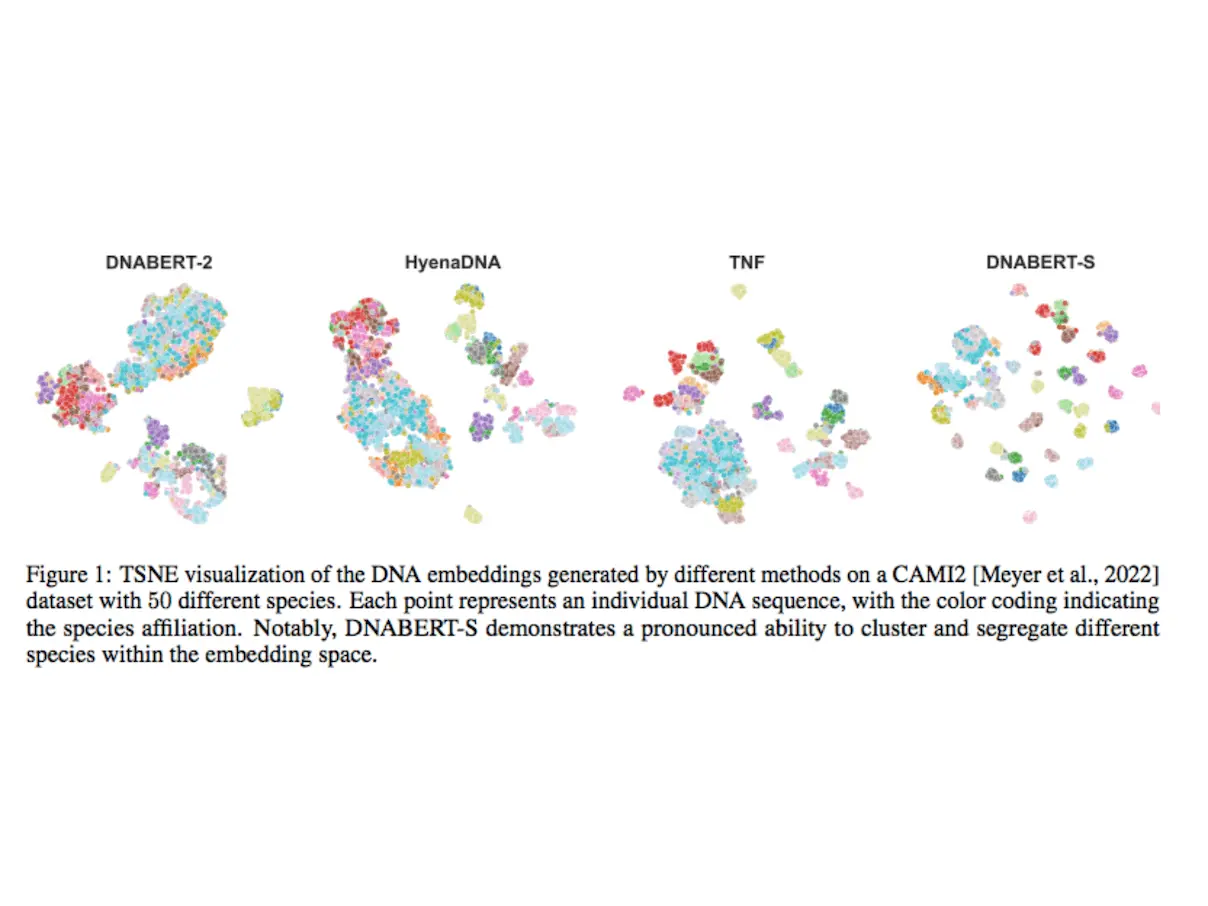
To address this challenge, researchers offer DNABERT-S, a genome foundation model designed to generate species-specific DNA embeddings. DNABERT-S outperforms other approaches by efficiently clustering and separating various species inside the embedding space. This enhanced performance stems from the proposed Manifold Instance Mixup (MI-Mix) loss and Curriculum Contrastive Learning (C2LR) strategy.
To evaluate DNABERT-S against existing methods, researchers compared DNABERT-S against other approaches and compiled a comprehensive benchmark that included more than 1,000 species, reflecting the diversity of natural microbial communities. This benchmark combines complicated datasets from CAMI2, a top metagenomics binning benchmark, with comprehensive reference genomes from Genbank. DNABERT-S delivers significant enhancements in DNA embedding, allowing for more in-depth and accurate genomic analysis. Its open-source nature encourages cooperation and creativity, pointing to a future in which genetic analysis leads to significant discoveries and better human health.
Introducing DNABERT-S: A Species-Aware Approach
DNABERT-S is based on two revolutionary techniques:
- Manifold Instance Mixup (MI-Mix)
This strategy smartly increases the virtual labels associated with each DNA sequence, creating a more robust learning environment for the model. This strong objective function promotes robust embeddings of error-prone sequences. It operates as follows:
- Adding hidden representations of DNA sequences to randomly selected levels inside the model’s architecture.
- In the output layer, train the model to detect and differentiate between these mixed representations.
This mixing procedure drives the model to learn invariant properties from DNA sequences, making it more resistant to noise and mistakes.
- Curriculum Contrastive Learning (C2LR)
This technique uses a well-planned learning schedule to gradually increase the difficulty of the tasks offered to the model. This method improves MI-Mix by progressively increasing the complexity of mixed representations. Initially, the model operates on basic combinations, allowing it to comprehend the fundamental notion. As it learns, the combinations get increasingly complex, requiring the model to improve its comprehension of subtle species-specific characteristics.
Unveiling Remarkable Findings
The researchers thoroughly evaluated DNABERT-S on 18 different datasets, and the findings speak for themselves:
Species classification
Despite having insufficient training data, DNABERT-S performed admirably in species categorization. In a 10-shot classification assignment, it outperformed the best baseline with only two shots of training! Grouping related species together improves precision, leading to a better understanding of biodiversity and evolutionary links.
Species clustering
The model excelled in classifying DNA sequences into species. DNABERT-S quadrupled the Adjusted Rand Index (ARI) over current models, exhibiting greater clustering accuracy. Identifying species from DNA sequences gets increasingly accurate, which helps in fields such as metagenomics and disease detection.
Metagenomics binning
Metagenomics binning is a technique for identifying species from complicated DNA combinations. DNABERT-S dramatically enhanced the number of properly-recognized species, providing the path for a greater understanding of microbial communities. Understanding the different microbial communities within an ecosystem becomes more efficient, benefiting environmental and health studies.
The Future of DNABERT-S
DNABERT-S enables more accurate and comprehensive genomic analysis in DNA embedding. Beyond early success, it can lead to exciting new possibilities:
Personalizing medicine
Customizing medicine entails developing more tailored drugs and therapies by studying individual genomes in more detail. Concentrating on specific problems allows for targeted and exceptionally successful solutions.
Understanding of complex diseases
Understanding complex disorders involves grasping the complex link between genes, species, and disease progression. This effort’s findings have the potential to drive further research in a range of genetic analytic domains.
Exploring unexplored territories
Exploring hidden microbial environments and discovering rare species with potential biotechnological applications are two instances of unexplored areas. Combining cutting-edge approaches has the potential to lead to significant scientific breakthroughs.
Publicly Available for Exploration
The DNABERT-S code, data, and pre-trained model are publicly available on GitHub to enable researchers to accelerate scientific advancement. This open-source approach invites scientists to explore its potential and contribute to its development.
Conclusion
Researchers introduced DNABERT-S, a novel genome foundation model designed to generate effective, species-aware DNA embeddings. To facilitate the training of DNABERT-S, they introduce the Manifold Instance Mixup (MI-Mix) training objective and the Curriculum Contrastive Learning (C2LR) strategy. They perform extensive experiments on 18 datasets across a variety of challenging tasks, including species clustering, classification, and metagenomics binning, to demonstrate the remarkable ability of DNABERT-S.
The species-aware training approach of DNABERT-S does not lead to better performance in unrelated genomic tasks such as human genome promoter prediction. The DNABERT-S will considerably aid species-related studies, such as identification, metagenomics binning, biodiversity assessment, and comprehension of evolutionary connections. However, the researcher’s observational findings offer helpful information for advancing the field of genomic application research.
Article source: Reference Paper
Important Note: arXiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Anchal is a consulting scientific writing intern at CBIRT with a passion for bioinformatics and its miracles. She is pursuing an MTech in Bioinformatics from Delhi Technological University, Delhi. Through engaging prose, she invites readers to explore the captivating world of bioinformatics, showcasing its groundbreaking contributions to understanding the mysteries of life. Besides science, she enjoys reading and painting.











 technique and Curriculum Contrastive Learning (C2LR).){kind=link}