
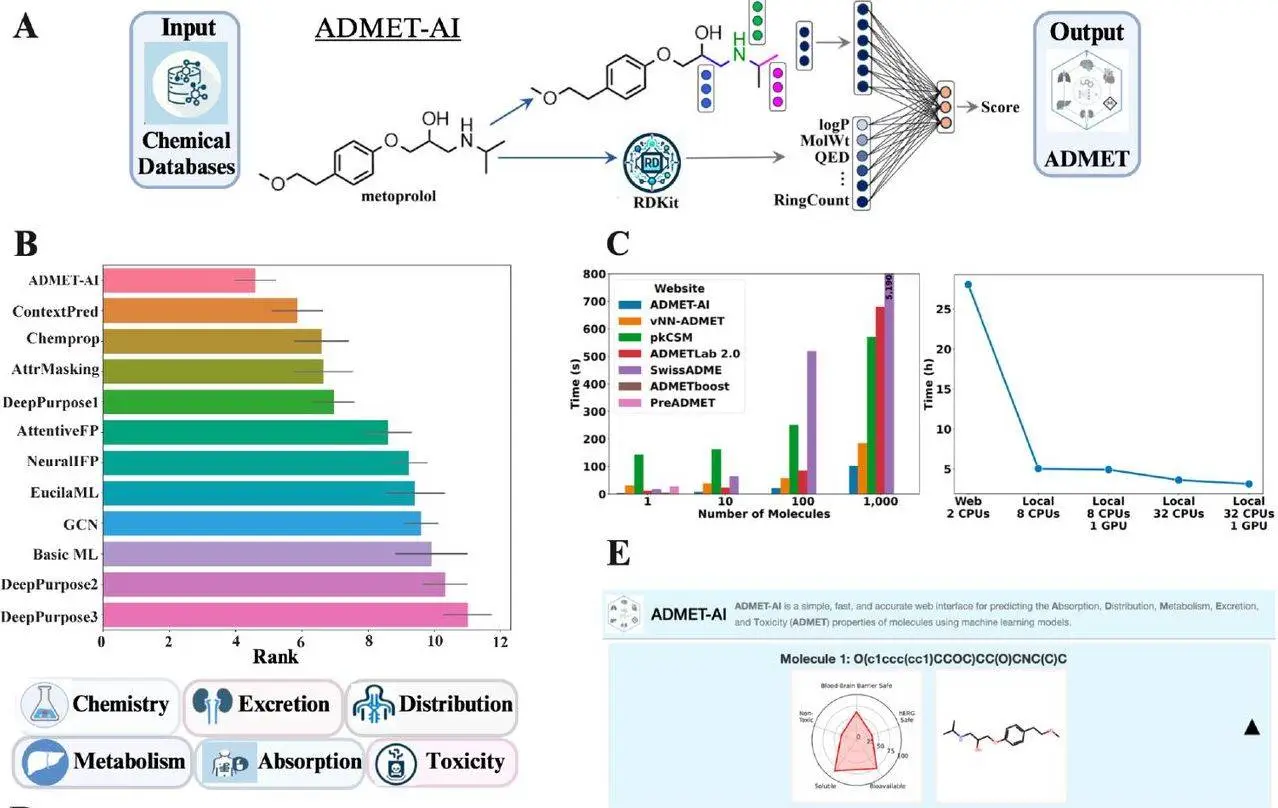
A vital step in drug discovery is accurately evaluating potential drug candidates’ Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET). Traditionally, this process has been time-consuming, expensive, and even limited to small compound libraries, making it difficult to test larger libraries. Recently, a group of researchers developed ADMET-AI, a simple, fast, and accurate platform for ADMET property prediction that includes both a web interface and a Python package for local prediction. It uses a graph neural network called Chemprop-RDKit, which was trained on 41 ADMET datasets from the Therapeutics Data Commons (TDC). It also surpasses existing ADMET prediction tools in terms of speed and accuracy while also providing many additional useful features. The best thing is it is free and open-source, making it a useful aid in the evaluation of large-scale chemical libraries for druglike compounds.
Introduction
ADMET is an important method to predict the behavior and potential side effects of the drug in our body. For a small molecule to be at the center of a therapeutic strategy and proceed from discovery to clinical trials, the compound must possess optimal ADMET properties. Scientists can identify potential drug interactions and other effects before the drug reaches the market and then can optimize its design for better efficacy and safety.
- Absorption: The rate and extent of absorption is influenced by the route of administration (e.g., oral, inhalation, injection, etc.), and the entrance of a drug into the bloodstream is the first step for any drug discovery and delivery. For example, an injected drug when injected enters the bloodstream directly, while an orally taken drug needs to be absorbed through the intestines.
- Distribution: Some factors, like the drug’s solubility, protein binding, and blood flow, will affect how it is distributed. There are some target-specific, and others may be distributed more widely throughout the body. Once the drug enters the bloodstream, it is distributed to different parts of the body, such as organs, tissues, etc.
- Metabolism: Metabolites are immediate products formed after metabolism, and some of them can be active or even toxic. The drug is chemically transformed into metabolites, facilitating its excretion, often making the drug more water-soluble. This process takes place in the liver and other organs.
- Excretion: Urine and feces are primary routes of excretion, but some drugs may also be excreted through sweat, breath, or breast milk. This is the final step, where the drug and its metabolites are eliminated from the body.
- Toxicity: The drug can potentially interact with other molecules or cause unwanted effects throughout the process; these effects are collectively referred to as toxicity. Toxicity can range from mild to life-threatening.
The success of the drug depends on its ADMET profile; thus, accurate evaluation of the profile is very important. Traditional methods like in vitro and in vivo assays lead to costly and lengthy processes in the drug discovery pipeline. Therefore, researchers used artificial intelligence (AI) to develop a powerful solution – ADMET-AI. It offers us a unique blend of accuracy, speed, and scalability, allowing researchers to analyze large-scale chemical libraries proficiently. It uses a deep learning model architecture for ADMET prediction called Chemprop-RDKit trained on diverse datasets that is 41 ADMET datasets (10 regression, 31 classification) from the Therapeutics Data Commons.ADMET-AI is freely available and is an open-source platform, making it an undefeatable tool.
ADMET-AI Model development
- Data: ADMET-AI uses 41 ADMET datasets (10 regression, 31 classification) from the Therapeutics Data Common (TDC, v0.4.1). Of those 41 datasets, 22 datasets (9 regression, 13 classification) form the TDC ADMET Benchmark Group leaderboard, which enables a side-by-side comparison of ADMET-AI compared to other ADMET prediction models. Two multi-task datasets were created, one containing all 10 regression datasets and one containing all 31 classification datasets, enabling the training of just two models (one regression, one classification) that cover all 41 ADMET properties.
- Model: ADMET-AI employs a Chemprop-RDKit deep learning architecture, combining a graph neural network (Chemprop) with 200 computed molecular features by RDKit (cheminformatics package). The Chemprop aggregated information through message passes to create a molecule representation by processing each atom and bond. The resulting representation, together with RDKit features, is fed through neural network layers for prediction.
- Training: Five models are trained with different train/validation/test splits for each dataset, and the average performance across the five test splits for that dataset is recorded. An ensemble prediction is averaged from these five models for each dataset at inference.
Advantages of ADMET-AI
- Benchmark Performance: It achieved the highest average rank among all models evaluated on the 22 datasets in the TDC ADMET Benchmark Group leaderboard. With its superior performance across various ADMET properties positions, ADMET-AI becomes an ideal tool for comprehensive ADMET analysis.
- Strong Performance Across Datasets: Single-task ADMET-AI models demonstrate strong performance, with R2 > 0.6 for 5 of the 10 regression datasets and AUROC > 0.85 for 20 of the 31 classification datasets.
- Speed: The prediction speed of the ADMET tool is very important. By outperforming other web servers in speed, which is 45% faster than the next best server for predicting ADMET properties of 1,000 molecules, ADMET-AI proved to be the best. Due to the significant speed advantages, multi-task models are deployed in the ADMET-AI web server for faster predictions.
- Flexibility: The ADMET-AI models can also be run locally for high-throughput ADMET prediction, helping researchers to handle large-scale datasets and integrate ADMET prediction seamlessly into their existing workflows. Both local and web-based servers add flexibility for users while addressing privacy concerns.
- Accuracy: ADMET-AI proved to be a promising tool for finding drug candidates with the optimal druglike profile. It delivers exceptionally accurate predictions across diverse ADMET properties.
ADMET-AI offers a curated set of approved drugs as a reference for contextualizing predicted ADMET properties, allowing users to select relevant subsets based on ATC codes for tailored comparisons. It displays results for 25 molecules with a download for the remaining ones while predicting up to 1,000 molecules at a time. It allows the user to select an appropriate subset of the DrugBank reference based on Anatomical Therapeutic Chemical (ATC) codes.
Conclusion
ADMET-AI marks a big advancement in ADMET evaluation by offering better speed, accuracy, and scalability for analyzing large-scale chemical libraries. Using a Chemprop-RDKit graph neural network for ADMET prediction is currently the most accurate model on average across the Therapeutics Data Commons ADMET leaderboard. The ADMET-AI website is the fastest web-based ADMET predictor, with a 45% reduction in time compared to the next fastest ADMET web server. In addition to the web interface, the ADMET-AI platform includes a Python package that can be applied locally as a command line tool for large-scale evaluation or as a Python module for use within other drug discovery tools.
The versatility of ADMET-AI holds promise in diverse fields, such as predicting the fate and toxicity of chemicals in environmental toxicology. The de novo creation of molecules with optimal ADMET profiles can be enabled by the integration of ADMET-AI with generative AI, which will revolutionize drug discovery. With its efficiency and accuracy, it will accelerate the development of more effective and much safer treatments and solutions.
Note: The ADMET-AI platform is freely available both as a web server and as an open-source Python package for local batch prediction (also archived on Zenodo). All data and models are archived on Zenodo.
Article Source: Reference Paper
Learn More:




Anchal is a consulting scientific writing intern at CBIRT with a passion for bioinformatics and its miracles. She is pursuing an MTech in Bioinformatics from Delhi Technological University, Delhi. Through engaging prose, she invites readers to explore the captivating world of bioinformatics, showcasing its groundbreaking contributions to understanding the mysteries of life. Besides science, she enjoys reading and painting.











{kind=link}