
Advances in high-throughput omics profiling have improved cancer patient classification significantly. However, insufficient data in multi-omics integration is a big problem because conventional techniques like imputation and sample exclusion frequently jeopardize biological connections and diversity. Moreover, it is sometimes overlooked how important it is to appropriately classify newly diagnosed patients with incomplete omics data into preexisting categories. Researchers from the University of Toronto, Canada, and their collaborators present IntegrAO (Integrate Any Omics), an unsupervised framework for integrating insufficient multi-omics data and categorizing novel samples. To create unified patient embeddings, IntegrAO first merges partially overlapping patient graphs from several omics sources and applies graph neural networks.
Introduction
Our genetic landscape is now painted in great depth by high-throughput omics technologies like transcriptomics and genomics, which provide previously unheard-of insights into health and disease. These technologies power patient stratification in the field of cancer, an essential procedure that divides patients into groups according to their distinct biological profiles. This, in turn, opens the door for precision oncology, in which individualized therapy regimens are designed for each patient. The road to customized treatment is rarely easy. A significant obstacle is the intrinsic incompleteness of omics data. Precision medicine and illness subtyping have made progress, but they are all limited by the need for comprehensive data across samples. This requirement becomes troublesome since missing data in profile assays is common, frequently the result of budgetary or experimental limitations.
Analyzing incomplete omics data is difficult. Excluding samples with missing omics data reduces the sample size, especially when integrating many layers. However, imputing missing values might create bias and uncertainty. The necessity for computational tools that can describe heterogeneous multi-omics datasets “as is” without requiring complete measurements or discarding relevant information is highlighted.
Challenges in multi-omics data integration
- Missing values make incomplete multi-omics data challenging to integrate.
- Current approaches struggle with incomplete data or require comprehensive data.
- It is difficult to classify newly diagnosed patients with incomplete data into preexisting subgroups.
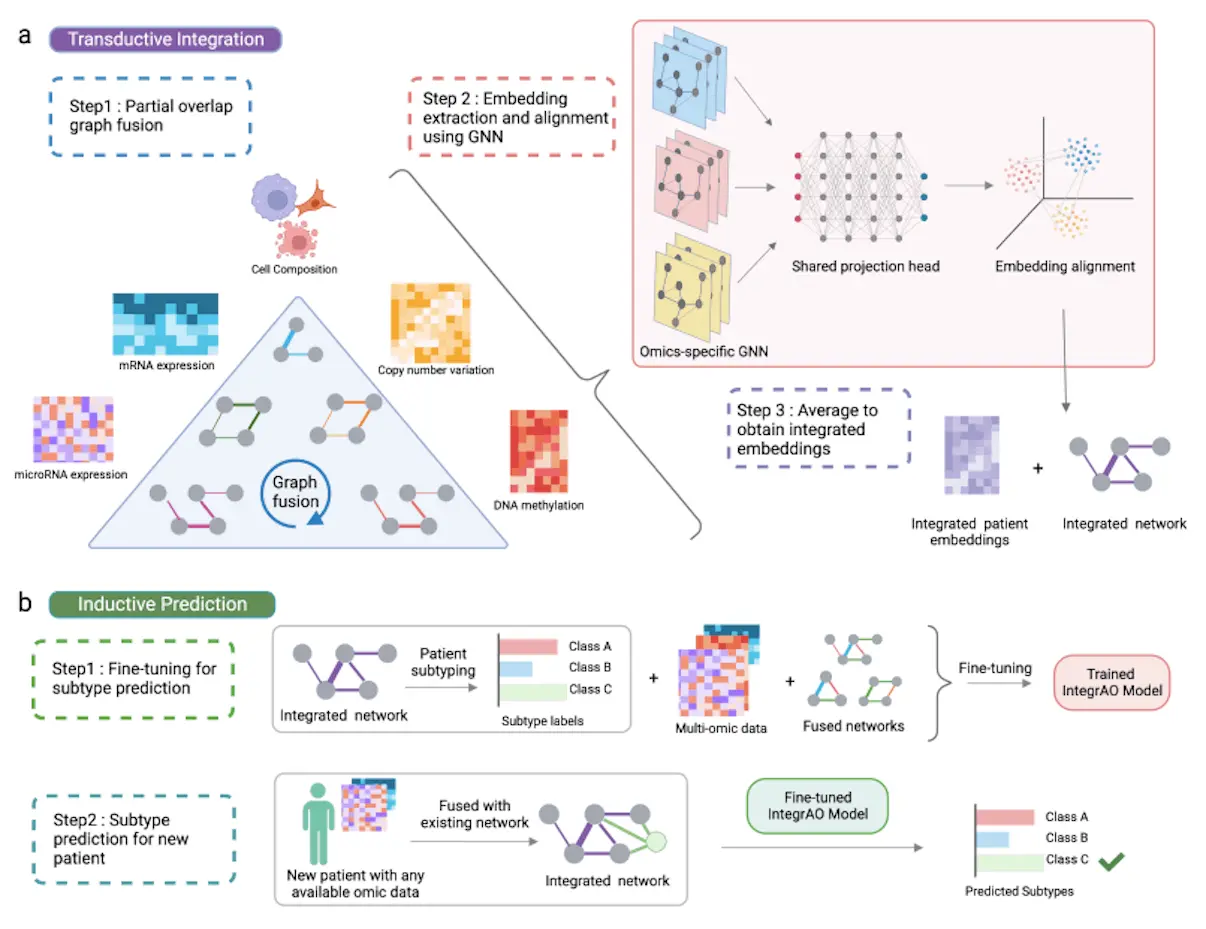
To overcome these challenges, a group of scientists came up with a solution called IntegrAO. It is an unsupervised framework that integrates incomplete multi-omics profiles and classifies fresh samples with incomplete data. It begins by integrating partially overlapping patient graphs from various omics data. The innovative partial graph fusion approach improves information integration for shared patients across several modalities and adjusts to conditions with fewer overlapping samples. It can also successfully incorporate partial omics data, resulting in excellent fidelity and noise resistance. The method uses graph neural networks (GNNs) to extract and align patient embeddings from various raw omics characteristics into a single space. The uniform embedding space is crucial for accurately categorizing new patients into predefined subtypes.
IntegrAO’s performance highlights
- Performance: Simulation experiments show that IntegrAO integrates partially missing data robustly, outperforming alternative approaches in both low and high-overlap cases.
- AML dataset: 12 different AML subtypes with distinctive biological traits, mutations, and survival traits are identified by IntegrAO. When compared to earlier classifications that relied just on cell hierarchy, these subtypes offer a finer resolution.
- Pan-cancer evaluation: Across five different cancer types, IntegrAO consistently finds subtypes with higher survival differentiation and clinical enrichment than other approaches.
- Classification of new patients: Even with incomplete omics data, IntegrAO performs noticeably better than other classifiers in placing new patients into preset subtypes. It is extremely helpful for clinical applications because of its resilience.
Methods
1. Data Preprocessing
Cancer Modelled Omics Datasets:
- The InterSim CRAN package across 15 clusters produced 500 samples.
- Setting hyperparameters for realistic clinical scenarios.
TCGA Databases for Cancer:
- cBioportal provided information on protein expression, DNA methylation, mRNA, copy number variation, and miRNA.
- Preprocessed data with normalization, imputation for missing values, and outlier reduction applied.
- Patients with a high rate of missing data or characteristics were excluded.
- The top 2,000 characteristics with the largest standard deviation were chosen for each modality.
AML Cancer Information Set:
- Combined information from the Leucegene, BEAT-AML, and TCGA cohorts.
- Variance-stabilizing transformation was used to normalize the gene expression data.
- Used the One Cell at a Time (OCAT) technique to perform the batch correction.
- OCAT was used to further reduce features.
- Over 2,000 very varied characteristics of DNA methylation.
- The final dataset comprised 308 patients with extra DNA methylation data and 812 AML patients with cell hierarchy and mRNA data.
2. Transductive Integration
Graph Fusion:
- Creates an omic (mRNA, DNA methylation, etc.) patient graph for each.
- Computes edge weights between patients using the Euclidean distance.
- Carries out two actions on every graph:
- The affinity matrix is normalized for numerical stability.
- Obtains the local affinity matrix by limiting the number of patients to the K most similar.
Partial Overlap Graph Fusion (for two modalities):
- Used common patients amongst modalities to spread data among graphs.
- Utilizing transition matrices, update the patient graph iteratively for every modality.
- Used a unique scaling normalization to accommodate varying numbers of typical patients.
- Updated the graphs of both modalities concurrently till convergence.
- Expanded on pairwise fusion techniques for multimodalities involving more than two data types.
Alignment and Extraction of Embeddings:
- Takes patient embeddings and aligns them across omics modalities.
- Makes use of two essential elements:
- Omic-specific graph encoders identify and get patient embeddings for every type of data.
- Map the embeddings from various omics into a common latent space using shared projection layers.
- To accomplish embedding alignment, the objective function minimizes both reconstruction loss and alignment loss.
- The final representation of the model is derived by averaging the patient embeddings across modalities.
3. Inductive Prediction
Model Fine-tuning for Subtype Prediction:
- Applied a prediction head to the unsupervised IntegrAO model to refine it.
- Determined classification loss to forecast new patient subtypes.
- Used trade-off settings for reconstruction, alignment, and classification losses to optimize the overall loss.
Subtype Prediction for New Patient:
- Added the omics data of the new patient to the data matrix and fusion matrix.
- Predicted the subtype probability distribution for the new patient using the refined IntegrAO model.
4. Cluster Number Selection:
- Used the Gaussian Mixture Model and log-likelihood scores to implement a certain strategy.
- Log-likelihood score means, and standard deviation for various cluster counts were computed.
- The ideal cluster number was found by deducting the mean from the standard deviation.
5. Gene Expression Deconvolution
- Created cell composition data for benchmarking using Bayes deconvolution.
- Using the Bayes deconvolution web portal’s default pretreatment processes and configurations.
- For integration benchmarking, use the generated cell composition matrices as the cell composition modality.
Key Features
- Missing modalities in a variety of cancer datasets are seamlessly integrated by IntegrAO.
- Even with incomplete profiles, it can classify new cases with reliability.
- In a variety of circumstances, IntegrAO is robust against missing data.
- It efficiently integrates several data formats to find clinically significant subgroups.
- In fresh sample projection and subtype identification, IntegrAO performs better than other approaches.
- For precision medicine, its capacity to manage missing data makes it indispensable.
- Personalized therapies and harmonized patient representations can be built with IntegrAO.
IntegrAO demonstrates the revolutionary potential of integrative analysis in precision oncology. There will come a time when all patients, regardless of any gaps in their omics profile, will be able to receive customized treatment programs due to their ability to handle partial data. In the end, IntegrAO can be very helpful in handling both complete and imperfect data, identifying clinically significant subcategories, and classifying new patients with partial data.
Conclusion
IntegrAO tries to solve two known problems in multi-omics analysis:
1. using partial profiles to project fresh samples and
2. insufficient heterogeneous data management.
The results validate IntegrAO’s ability to integrate different cancer datasets with missing data and accurately classify new patients. Testing with simulated cancer omics data showed the ability to integrate missing data under different conditions, proving that the system performs well for bigger overlaps and is resilient against noise at smaller data overlaps. IntegrAO effectively combined transcriptomics, DNA methylation, and cell hierarchy composition, leading to the identification of 12 physiologically and clinically distinct subgroups and illuminating the heterogeneity of AML in the acute myeloid leukemia case study.
IntegrAO is the answer to achieving the full potential of precision medicine. It acknowledges the complexity of biological data and draws links between s unconnected elements. Its features enable tailored diagnosis, therapy selection, and prognosis prediction. It has even been shown to be a potent method for cancer subtype prediction and multi-omics integration. It helps handle both complete and incomplete data, showing its superiority over the other traditional methods and proving its potential to be a revolutionary tool.
Article Source: Reference Paper | The code to utilize IntegrAO is available on GitHub
Important Note: arXiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Anchal is a consulting scientific writing intern at CBIRT with a passion for bioinformatics and its miracles. She is pursuing an MTech in Bioinformatics from Delhi Technological University, Delhi. Through engaging prose, she invites readers to explore the captivating world of bioinformatics, showcasing its groundbreaking contributions to understanding the mysteries of life. Besides science, she enjoys reading and painting.











{kind=link}