
The drug development process involves a winding trail across the enormous chemical wilderness, looking for compounds that interact ideally with their target proteins. Traditional approaches, while successful, frequently stumble in the dark, unable to capture the delicate 3D pattern of particles and their interactions. But do not worry; scientists from Beijing StoneWise Technology Co Ltd, China, found a solution. Lingo3DMol is a groundbreaking technology that combines the power of language models with geometric deep learning to overcome the limits of previous approaches. In Lingo3DMol, language and geometry work together to shape molecules into intricate 3D architectures nestled within their protein partners rather than flat lines on paper.
Introduction
Structure-based drug design entails creating compounds that can bind to a specific target protein, which is an important and difficult drug development process. De novo chemical production utilizing artificial intelligence has lately acquired popularity as a technique for drug development. Earlier molecular generative models used either molecular string representations or graph representations. However, both representations ignore three-dimensional (3D) spatial interactions, making them unsuitable for target-aware molecule creation. The increased availability of 3D protein-ligand complex structure data, combined with breakthroughs in geometric deep learning, has enabled artificial intelligence systems to design molecules with 3D binding poses directly. For example, approaches using 3D convolutional neural networks are utilized to capture 3D inductive bias. However, they still fail to transform atomic density grids into discrete molecules.
While graph-based 3D molecular generation approaches have lately shown considerable promise, they still struggle to reproduce reference molecules in a particular pocket without information leakage, which is a crucial baseline for evaluation. To address these challenges, researchers introduced Lingo3DMol, a pocket-based 3D molecule generation method that combines language models and geometric deep learning technology.
Challenges in 3D Molecule Generation
- Existing techniques using string or graph representations lack 3D knowledge, resulting in undesired molecules.
- Autoregressive generation methods can become stuck in local optima, accumulating mistakes.
- Difficulty replicating reference molecules without information loss.
Lingo3DMol breaks free from the constraints of typical 2D representations, adopting a revolutionary “fragment-based SMILES” language that encodes both molecular bonds and atomic locations in magnificent 3D. This allows the model to understand the spatial dance of atoms rather than merely their linear relationships. It does not overlook the important interaction with the target protein. It uses a specific non-covalent interaction predictor, similar to a competent matchmaker, to ensure that the created molecules fit snuggly into their respective pockets. No more getting lost in dead ends. Lingo3DMol also effectively navigates the enormous chemical space, avoiding unwanted or impossible molecule formations. It’s like having a GPS in the drug design world, guaranteeing you arrive at your goal (a potent, drug-like molecule) quickly and effectively.
Developing novel drugs is a time-consuming and expensive process that is frequently hampered by traditional approaches that fail to capture the critical 3D interactions between compounds and their target proteins. Lingo3DMol is a novel technique that tackles these restrictions through:
- Combining the capabilities of linguistic models with geometric deep learning: Lingo3DMol outperforms simpler representations (2D strings or graphs) by embedding 3D context directly into the language model, resulting in more accurate and realistic molecule synthesis.
- Understanding the 3D binding pockets: Lingo3DMol creates compounds that fit better by taking into account probable interaction locations within the protein pocket, potentially increasing medicinal efficacy.
- Overcoming limitations of existing technologies: Unlike certain autoregressive methods that produce unwanted structures, Lingo3DMol reduces the synthesis of unrealistic molecules while prioritizing drug-like features.
- Faster generation speed: When compared to other approaches, Lingo3DMol can create possible drug candidates much faster, significantly accelerating the drug development process.
Methods
Lingo3DMol transforms 3D drug creation by merging the power of language models and geometric deep learning. Here’s a breakdown of its main methodological features.
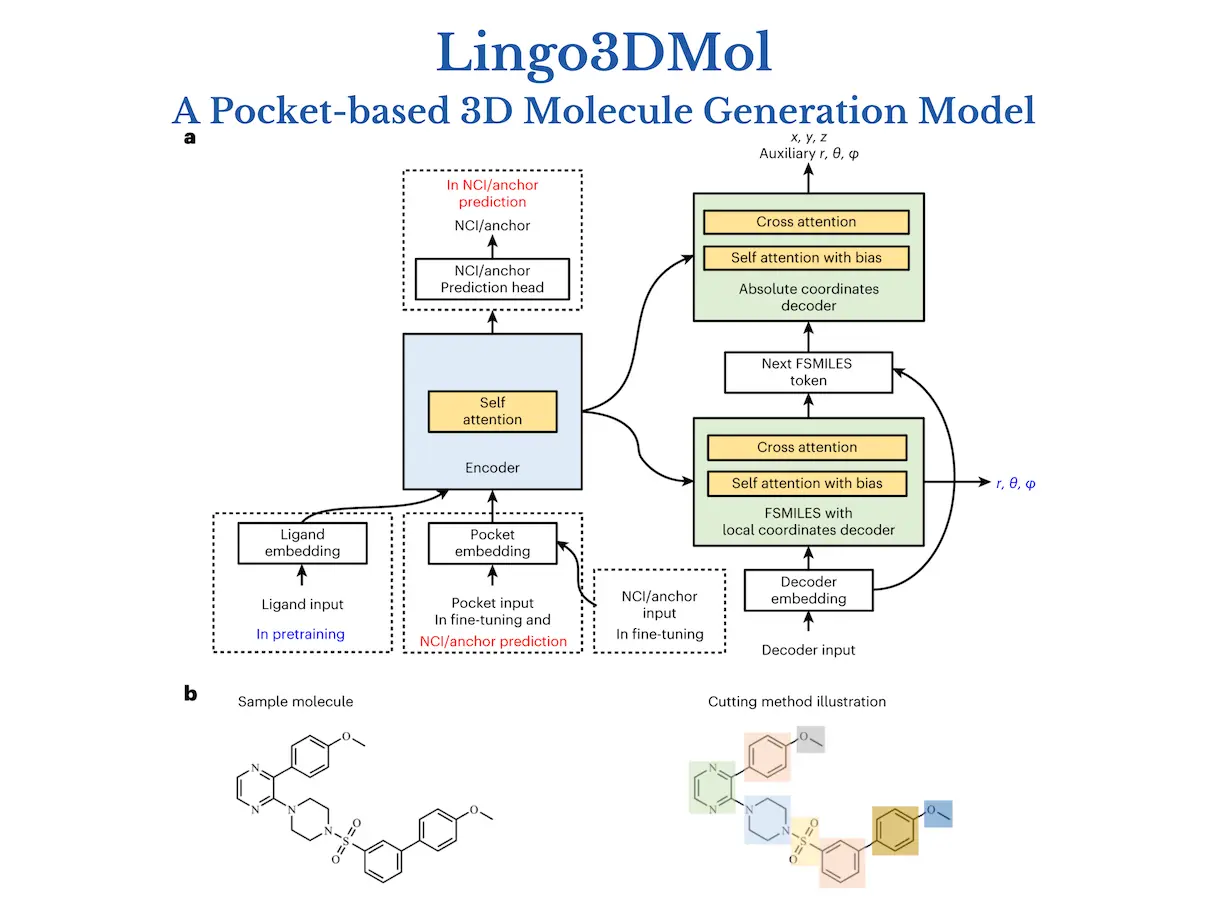
1. The Fragment-Based Simplified Molecular-Input Line Entry System (FSMILES):
- Traditional SMILES notation struggles to capture 3D context.
- Lingo3DMol introduces FSMILES, which encodes ring size in all ring tokens to provide better context during molecule formation.
- Ring-first traversal prioritizes ring creation, which results in more realistic topologies.
2. Combined Coordinate System:
- Accurately displaying both local and global 3D information is critical.
- Lingo3DMol uses a mixed coordinate system.
- Local spherical coordinates: Determine specific bond lengths and angles inside fragments.
- Global Euclidean coordinates describe the overall spatial arrangement of atoms.
3. Non-Covalent Interaction (NCI) or Anchor Predictor:
- Understanding the binding patterns between ligands and proteins is critical for good drug design.
- Lingo3DMol employs a distinct NCI/anchor predictor trained on ligand-protein complexes.
- This model reveals probable NCI binding sites on the target protein to help guide compound synthesis.
4. 3D Molecule Denoising Pretraining:
- Limited training data may impede model generalization.
- Lingo3DMol uses 3D molecule denoising pretraining, similar to BART and Chemformer, to improve its ability to handle unseen data.
- This increases the model’s robustness and adaptability to different target proteins.
5. Fine-tuning and evaluation
- Lingo3DMol was optimized using the PDBind2020 dataset of protein-ligand complexes.
- Its performance was evaluated on the tough DUD-E dataset and compared to cutting-edge approaches.
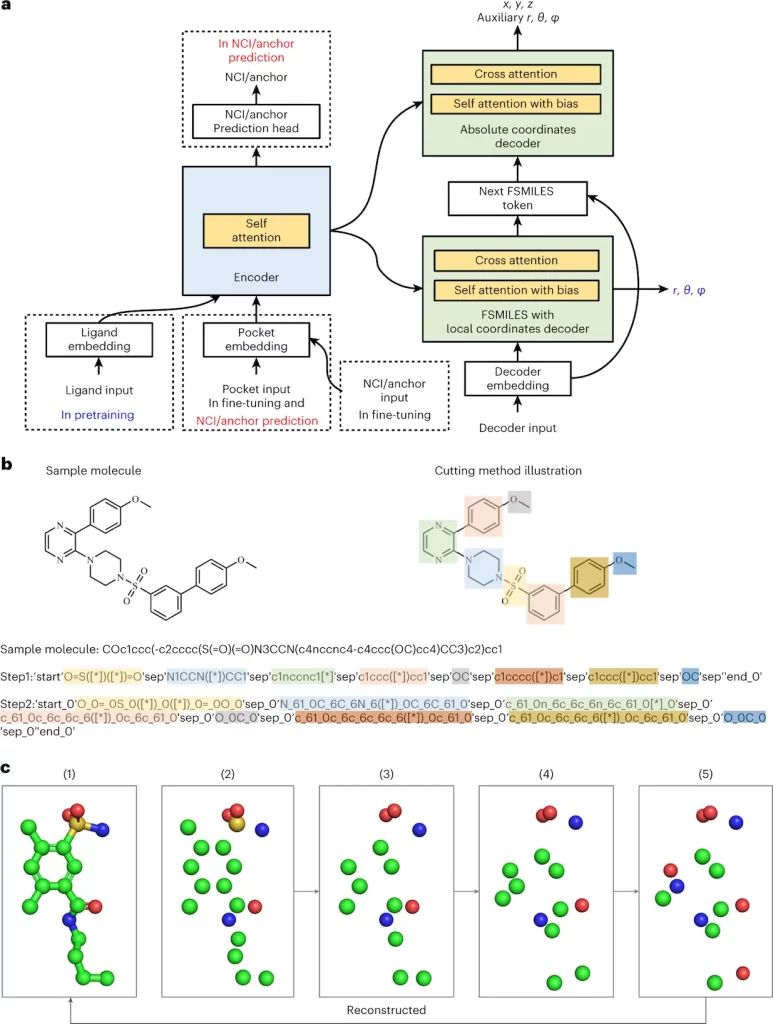
Image Source: https://doi.org/10.1038/s42256-023-00775-6
Comparison with Different Methods:
- TargetDiff, while providing non-autoregressive generation, nonetheless produces a significant number of unattractive structures.
- Graph-based approaches fail to replicate reference molecules without information leaking, which Lingo3DMol addresses.
- Lingo3DMol routinely outperforms SOTA approaches in a variety of criteria, indicating that it is a reliable and successful solution for 3D molecule production.
Lingo3DMol’s novel combination of inventive representations, pretraining techniques, and focused NCI prediction ushers in a new era of precise and economical three-dimensional drug development.
Challenges
Capturing all NCIs: The autoregressive generation process in Lingo3DMol makes it difficult to evaluate all potential non-covalent interactions (NCIs) within a single molecule. Exploring alternate representations, such as electron densities, that depict a more diverse interaction scene could provide a solution.
Achieving equivariance: A molecule’s rotational and translational fluctuations should not influence its anticipated properties. While Lingo3DMol uses augmentation and SE(3) invariant features, more research into current equivariant models like GVP and Vector Neurons should increase efficiency.
Comprehensive drug-like property assessment: Assessing drug-like qualities merely using case analysis and technologies such as QED and SAS is insufficient. A more systematic and complete examination is required, taking into account a wide range of features and possible adverse effects.
Conclusion
Capturing all of the delicate interaction within the protein pocket is still a problem, but Lingo3DMol, ever the inventor, is already working on solutions. Exploring electron density representations, a more complex tapestry of interconnections than just atomic positions has enormous promise. And, like a smooth dancer unaffected by pirouettes and translations, Lingo3DMol uses rotation and translation augmentation to stay grounded while utilizing SE(3) invariant features such as distance matrices and local coordinates to waltz with any molecule, regardless of orientation.
However, issues remain. Capturing all NCIs within a single molecule is difficult due to the autoregressive production process, and researchers intend to examine this more. Representing molecules and intermolecular interactions with electron densities appears to be a viable direction, and some related research may serve as a suitable starting point. Furthermore, the equivariance trait is an important aspect of 3D molecule production. There are numerous studies on rotational and translational equivariant models, including GVP and Vector Neurons. Scientists use rotation and translation augmentation to improve the model and SE(3) invariant features such as distance matrices and local coordinates to relieve the problem.
Lingo3DMol potential is as large as the chemical space itself. From tailored medicine to defeating previously formidable opponents, it opens the door to previously unthinkable medical discoveries. It is rewriting the story of drug creation, one 3D masterpiece at a time.
Article Source: Reference Paper | The source code of Lingo3DMol is available on GitHub | An online service is available at https://sw3dmg.stonewise.cn
Follow Us
Learn More:




Anchal is a consulting scientific writing intern at CBIRT with a passion for bioinformatics and its miracles. She is pursuing an MTech in Bioinformatics from Delhi Technological University, Delhi. Through engaging prose, she invites readers to explore the captivating world of bioinformatics, showcasing its groundbreaking contributions to understanding the mysteries of life. Besides science, she enjoys reading and painting.











{kind=link}