
Molecular geometry modeling, which involves building models that show the spatial arrangement of atoms in a molecule, usually helps with this understanding. The field of molecular modeling has been transformed by geometric deep learning. However, due to molecules’ inherent 3D shape and the intricate interactions between their constituent atoms, standard deep learning algorithms need help to solve this challenge. While the current generation of neural network models approaches ab initio accuracy for molecular property prediction, high computing costs, and insufficient geometric information usage have hampered their use in drug development and molecular dynamics (MD) modeling. Microsoft researchers introduced ViSNet, an equivariant geometry-enhanced graph neural network that predicts molecule structures with minimum processing costs while elegantly extracting geometric features.
Challenges in Traditional Deep Learning for Molecular Modeling
Modern research and engineering rely largely on molecular modeling, which fosters scientific and technological discoveries, helps understand chemical interactions, and facilitates the identification of innovative medications. On the one hand, group representation theory algorithms are entirely mathematical and make extensive use of geometric information via high-order geometric tensors. These algorithms, however, are frequently ineffective for big molecular systems, such as chemical and biological molecules, that lack periodic boundary constraints; rather, they may be suited for periodic systems with sophisticated model designs. This is because these algorithms frequently necessitate computationally costly procedures, such as the Clebsch-Gordan product (CG-product). Conventional deep learning algorithms frequently depict molecules using 2D grids or fixed-length vectors, omitting significant information that may be discovered in the 3D spatial arrangement of atoms and interactions. This complicates molecular dynamics simulations and leads to inaccurate predictions of different molecule properties.
Despite significant recent strides in geometry deep learning, however, challenges persist. These include:
- Insufficient molecular interpretability
- Rapidly increasing computing costs as molecular size increases
- Need for blind tests and evaluations in real applications
Structure-activity relationships (SAR), which are intricate interactions between molecular structure and biological function, can be better understood through the use of molecular geometry modeling. Fundamental to SAR is the idea that a molecule’s unique chemical structure—which encompasses not only the bonds between its nuclei but also its three-dimensional twists and structures—determines its biological activity. The ultimate objective of SAR is to predict the effects of molecular configurations on important processes, including drug interactions, chemical reactivity, and protein functioning. If this were possible, researchers might predict a drug’s toxicity, side effects, and effectiveness far before human testing.
ViSNet: A Novel Approach to Molecular Geometry Modeling
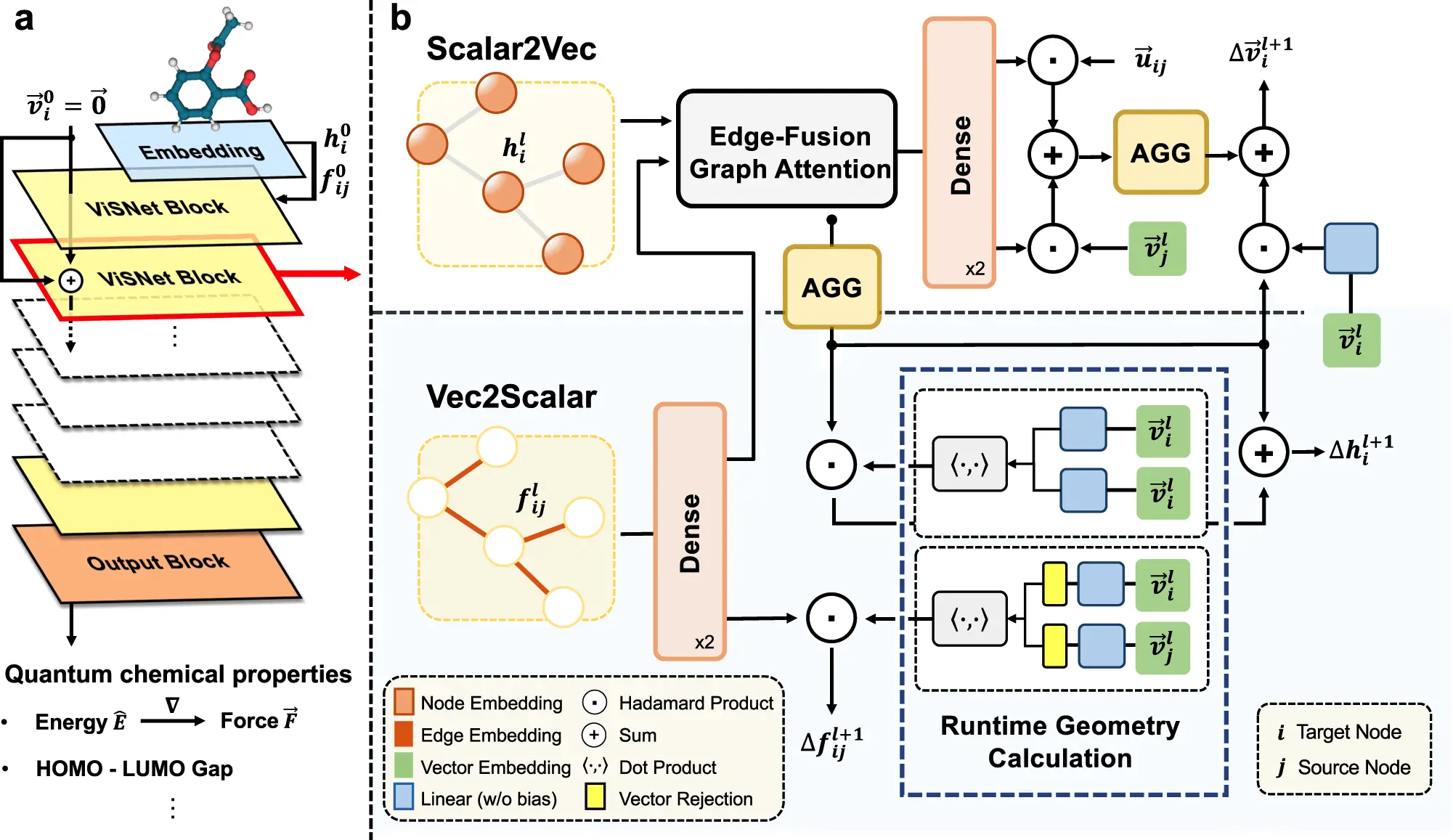
Conventional molecular dynamics (MD) simulations trace molecule movements using dihedral angles, bond length, and bond angle. The creators demonstrated a novel approach called the vector-scalar interactive graph neural network (ViSNet), which was influenced by these concepts and balances computational costs with geometric information utilization. ViSNet greatly accelerates model training and inference while consuming less memory by implicitly extracting different geometric characteristics, such as angles, dihedral torsion angles, and incorrect angles, by the force field of classical MD with linear time complexity.
Rather than just adding bond angle and dihedral data to our model, the creator introduced the concept of “direction units.” These units compute normalized vectors pointing from the center node to its neighboring nodes to represent nodes in the chemical structure as vectors. To account for interactions involving two-body, three-body, and quadruplet atoms, the researchers expanded the usual estimates of bond length, bond angle, and dihedral angles. To manage these interactions efficiently, they developed a runtime geometry calculation (RGC) module that accurately represents the complex interatomic linkages in molecules. Remarkably, the three- and four-body interaction calculations of the RGC module exhibit linear time complexity, guaranteeing computational efficiency.
Researchers employed the inner product to simplify the computationally intensive Clebsch-Gordan product and incorporate spherical harmonics into the vector form. Furthermore, they provided a well-thought-out vector-scalar interactive equivariant message passing (ViS-MP) approach that interacts scalar hidden representations with vector ones to fully exploit the geometric properties.
ViSNet outperforms all cutting-edge algorithms on all compounds in the MD17, revised MD17, and MD22 datasets when properly evaluated on a few benchmark datasets. It also outperforms the QM9 Molecule3D dataset, exhibiting the full capability of molecular geometric representation. ViSNet also won the OGB-LCS@NeurIPS2022 competition at the PCQM4Mv2 track.
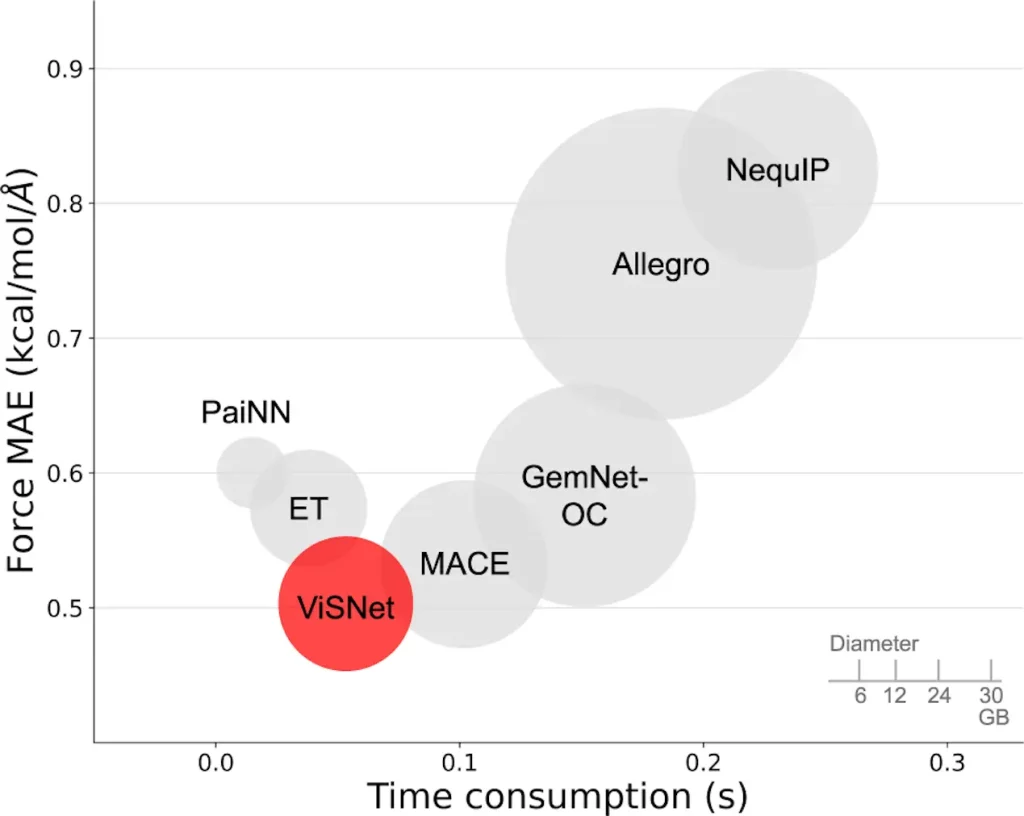
Image Source: https://doi.org/10.1038/s41467-023-43720-2
ViSNet’s Real-world Applications for Molecular Modeling and Property Prediction
To assess ViSNet’s practical value, researchers systematically tested its performance against recognized criteria for predicting molecular characteristics. Across various datasets, including MD17, updated MD17, MD22, QM9, and Molecule3D, ViSNet consistently outperformed current techniques, demonstrating outstanding accuracy in describing molecular geometry. Performance gains came from ViSNet’s integration of more geometric data and its complete utilization of geometric data in ViS-MP.
Next, they assess ViSNet using molecular dynamics (MD) simulations to predict the Chignolin protein’s function. To evaluate ViSNet as the potential for MD simulations, they incorporated ViSNet trained only with 950 samples on MD17 into the ASE simulation framework to perform MD simulations for all seven kinds of organic molecules. All simulations are run with a time step τ = 0.5 fs under the Berendsen thermostat with the other settings the same as those of the MD17 dataset. ViSNet outperformed conventional empirical force fields and has shown potential compared to current machine-learning force fields. ViSNet was trained using the AIMD-Chig dataset, which contained protein data processed with advanced density functional theory (DFT) techniques. Surprisingly, findings from extensive DFT calculations were almost duplicated in ViSNet simulations, revealing the technology’s potential for precise and effective data simulations.
In the First Global AI Drug Development Competition, scientists used ViSNet to compete in an international competition to predict inhibitors of the major protease of SARS-CoV-2 using small molecule sequence information (SMILES). One thousand one hundred five players from 878 teams worldwide participated in the competition. ViSNet demonstrated its great prediction accuracy, which helped it to win the competition.
Microsoft has included ViSNet as a fundamental model for molecular modeling and property prediction in PyTorch Geometric Library as a core model for molecular modeling and property prediction. Additionally, to guarantee continuous maintenance and refinement, users may now access the most recent version of ViSNet on GitHub.
Conclusion
Researchers suggest using a vector-scalar interactive graph neural network, or ViSNet, to address the issue of balancing the use of geometric information with computational costs. ViSNet greatly accelerates model training and inference while consuming less memory by implicitly extracting different geometric characteristics, such as angles, dihedral torsion angles, and incorrect angles, by the force field of classical MD with linear time complexity. They use spherical harmonics to enlarge the vector representation and substitute the inner product for the computationally expensive Clebsch-Gordan product. ViSNet efficiently navigates conformational space, offering interpretability by mapping geometric representations to molecular structures in simulations and case studies. Showing that it has great potential for precise and effective data simulations.
Article source: Reference Paper | Reference Article | The code used to reproduce the results is available on GitHub
Follow Us!
Learn More:




Anchal is a consulting scientific writing intern at CBIRT with a passion for bioinformatics and its miracles. She is pursuing an MTech in Bioinformatics from Delhi Technological University, Delhi. Through engaging prose, she invites readers to explore the captivating world of bioinformatics, showcasing its groundbreaking contributions to understanding the mysteries of life. Besides science, she enjoys reading and painting.











{kind=link}