
One must understand proteins’ structural flexibility in-depth to comprehend their role in biological processes and functional systems. It is still challenging to predict various conformational states and motions of proteins despite the advances in high-throughput technologies that have increased our understanding of protein structures. For a methodical and thorough explanation of the conformational diversity of protein families, researchers provide us with a Dimensionality Analysis for Protein Conformational Exploration (DANCE). It may support expected as well as experimental structures. It may be used to analyze anything, including superfamilies and individual proteins.
Unveiling the Dynamic World of Proteins
Proteins regulate all biological processes, and protein irregularities are frequent causes of illness. In recent years, high-throughput technologies have contributed to our understanding of their 3D structures and amino acid sequencing. These advancements have pushed the single-structure frontier, but they have also highlighted the complicated ways proteins move and alter to fulfill biological functions. New methods for studying protein conformation have been made possible by the development of AlphaFold, a neural network for predicting protein structure. Using state-annotated templates, modifying the depth and content of the input multiple sequence alignment, or forcing sampling are some possible approaches. Although extensive investigations have discovered constraints, intriguing findings have been shown for particular protein families. Research utilizing low-dimensional manifolds or representations to learn from simulated or observed conformations emphasizes how difficult it is to predict unknown states and how important it is to have high-quality training and benchmarking data.
DANCE: A Computational Breakthrough
Scientists present DANCE, a unique computational pipeline for maintaining and studying the many structural states of proteins. DANCE captures both linear and nonlinear motions by applying principal component analysis together with other methods, offering a comprehensive picture of protein flexibility. They highlighted the important findings, including the ability to evaluate distant homologs, quantify variability with few motions, and create plausible conformations. Severe conformational changes and reference reliance constraints are also discussed.
DANCE can handle structures with different amino acid sequences, both anticipated and experimental. It builds the conformational collections with objectivity, eschewing predefined protein or domain definitions. A detailed analysis of inter-domain motions is made possible by taking into account the entire context of input protein chains. Moreover, DANCE does not assume possible conformations in order to account for ambiguity arising from unclear protein regions. It presents a weighting system to lessen the uneven distribution of variable coverage.
A Glimpse into the Inner Workings of DANCE
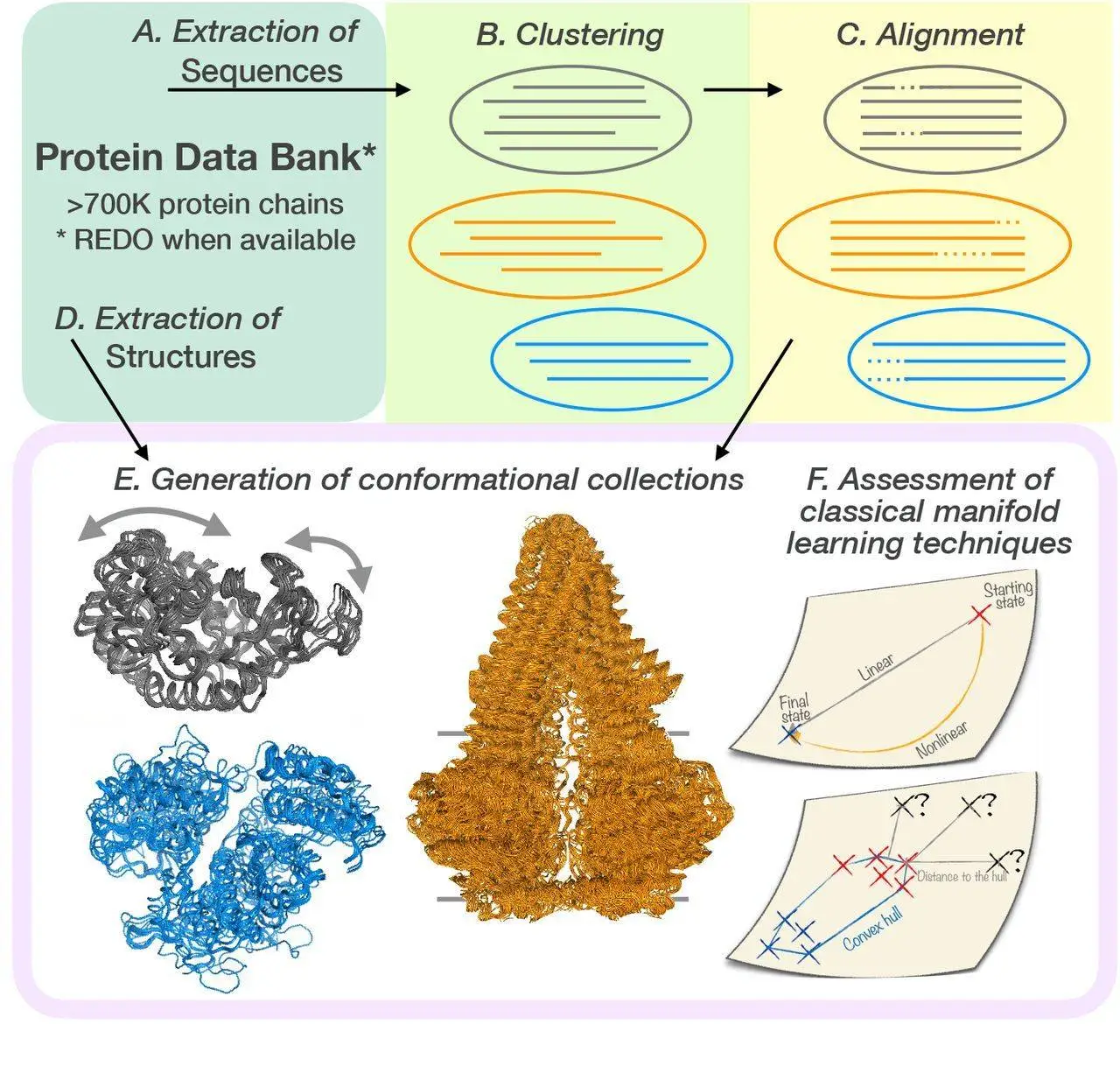
DANCE generates a set of conformational collections or ensembles that are unique to a certain protein or protein family when given a set of protein 3D structures in the Crystallographic Information File, or CIF, format (in CIF of PDB format). It first clusters and superimposes the input structures based on the similarities found in their corresponding amino acid sequences. It then determines the set of principal components sufficient to explain the variability observed within each conformational ensemble.
A more detailed explanation of the methods used in DANCE, along with some key points, are as follows:
Sequence-based clustering
- Method: MMseqs2 is a technique for grouping protein sequences based on how similar they are. MMseqs2 is a sensitive and fast sequence search tool that works by comparing profiles.
- Key points:
- The user can specify the coverage and degree of sequence similarity for grouping.
- This step helps the assembly of proteins with similar structures and activities.
Multiple sequence alignment
- Method: sequences within each cluster are aligned using MAFFT. A popular multiple-sequence alignment method that is accurate and effective is called MAFFT.
- Key points:
- The alignment shows regions necessary for structure and function as well as conserved residues.
- A tutorial for superimposing 3D structures is also included.
Structure extraction and alignment
- Method: DANCE uses protein structures to extract 3D coordinates for backbone atoms (N, C, Cα, and O). It then applies the ultrafast Quaternion Characteristic Polynomial approach to superimpose these structures based on sequence alignment. This approach reduces the root mean square deviation (RMSD) between structures.
- Key points: “X” symbols are used to reconstruct or substitute missing residues. This stage establishes a common reference frame for all structures in a cluster, allowing their conformations to be compared.
Conformational ensemble generation
- Method: The overlaid structures are organized into conformational ensembles based on sequence groupings.
- Key points: Each ensemble consists of comparable protein conformations. This stage analyzes protein flexibility and conformational diversity among related proteins.
Principal component analysis (PCA)
- Method: Based on each ensemble’s 3D coordinates, DANCE employs PCA to identify the principal components (PCs) of motion for that ensemble. In data, PCs are orthogonal paths that maximize variation.
- Key points: PCs may be used to investigate the flexibility of proteins, evaluate how different proteins move, and design new protein conformations. The most important protein motions often occur in the first few PCs.
DANCE’s Advantages
- Versatile and supports both experimental and anticipated structures.
- Unbiased, avoiding predefined protein or domain definitions.
- Considers whole protein chains for complete analysis.
- Handles uncertainty in unsettled regions.
- Provides databases of collections and benchmarks for study.
- Open-source code is accessible for wider use.
The future of the protein DANCE
DANCE is still growing, but it has enormous potential! Researchers are exploring:
- Multiple stages: Rather than a single reference, numerous structures are used to present a more comprehensive view.
- Ditching the stage: Creating reference-free approaches that allow proteins to move freely in the computational realm.
- Embracing uncertainty: Accounting for the inherent uncertainties in protein data allows for more nuanced interpretations.
Conclusion
DANCE has pirouetted into the scientific scene, providing a thrilling viewpoint on proteins’ dynamic repertory. By deconstructing their complex “steps” using principal component analysis and other approaches, it sheds light on the language of protein motion, revealing function secrets and opening the road for new therapies.
DANCE was developed by academics primarily to deal with single polypeptide chains arranged based on sequence similarity. The researchers have presented a proof-of-concept application study to demonstrate DANCE’s ability to appropriately characterize continuous motions shared by distant homologs with varied quantities of chains. This approach offers a multi-view perspective on a given collection of conformations, easing interpretability and allowing for augmenting data in a learning context. Future improvements will explore multi-reference or reference-free probabilistic frameworks and more refined accounts of data uncertainty.
Article source: Reference Paper | DANCE’s source code is available on GitHub
Follow Us!
Learn More:




Anchal is a consulting scientific writing intern at CBIRT with a passion for bioinformatics and its miracles. She is pursuing an MTech in Bioinformatics from Delhi Technological University, Delhi. Through engaging prose, she invites readers to explore the captivating world of bioinformatics, showcasing its groundbreaking contributions to understanding the mysteries of life. Besides science, she enjoys reading and painting.











{kind=link}