
Genesis Molecular AI and NVIDIA have introduced PEARL (Placing Every Atom in the Right Location), a deep learning foundation model designed for large-scale protein–ligand cofolding in computational drug discovery. By leveraging synthetic training data, SO(3)-equivariant diffusion architectures, and controllable inference, PEARL achieves more accurate and physically valid binding pose predictions, outperforming AlphaFold 3 and other leading approaches, and marking a significant advance in structure-based drug design.
Why Protein–Ligand Poses Matter
An AI model that can predict how a small molecule binds in a pocket of a protein right down to the angstrom level is a deceptively simple question. Active site binding is the cornerstone of structure-based drug design. In such cases, chemists modify the binding ligands to improve the potency, selectivity, and safety of the drug candidate. Induced-fit docking in docking software treats the protein as a rigid structure. Prior AI modeling approaches in deep learning either handled protein-only scenarios or offered a model that, while inceptively plausible, contained significant pose artifacts.
Meet Pearl: A Generative Foundation Model
Pearl is introduced as a generative “cofolding” foundation model that predicts full protein–ligand complexes in 3D, rather than docking a ligand into a fixed pocket. The model combines a transformer trunk with an SO(3)-equivariant diffusion module that natively respects 3D rotational symmetries, so rotating the input system corresponds to a rotated output, not a different prediction. This geometric bias, implemented via equivariant transformer blocks, enhances sample efficiency and enables the model to learn binding rules rather than memorizing specific examples.
Beating AlphaFold 3 Where It Hurts
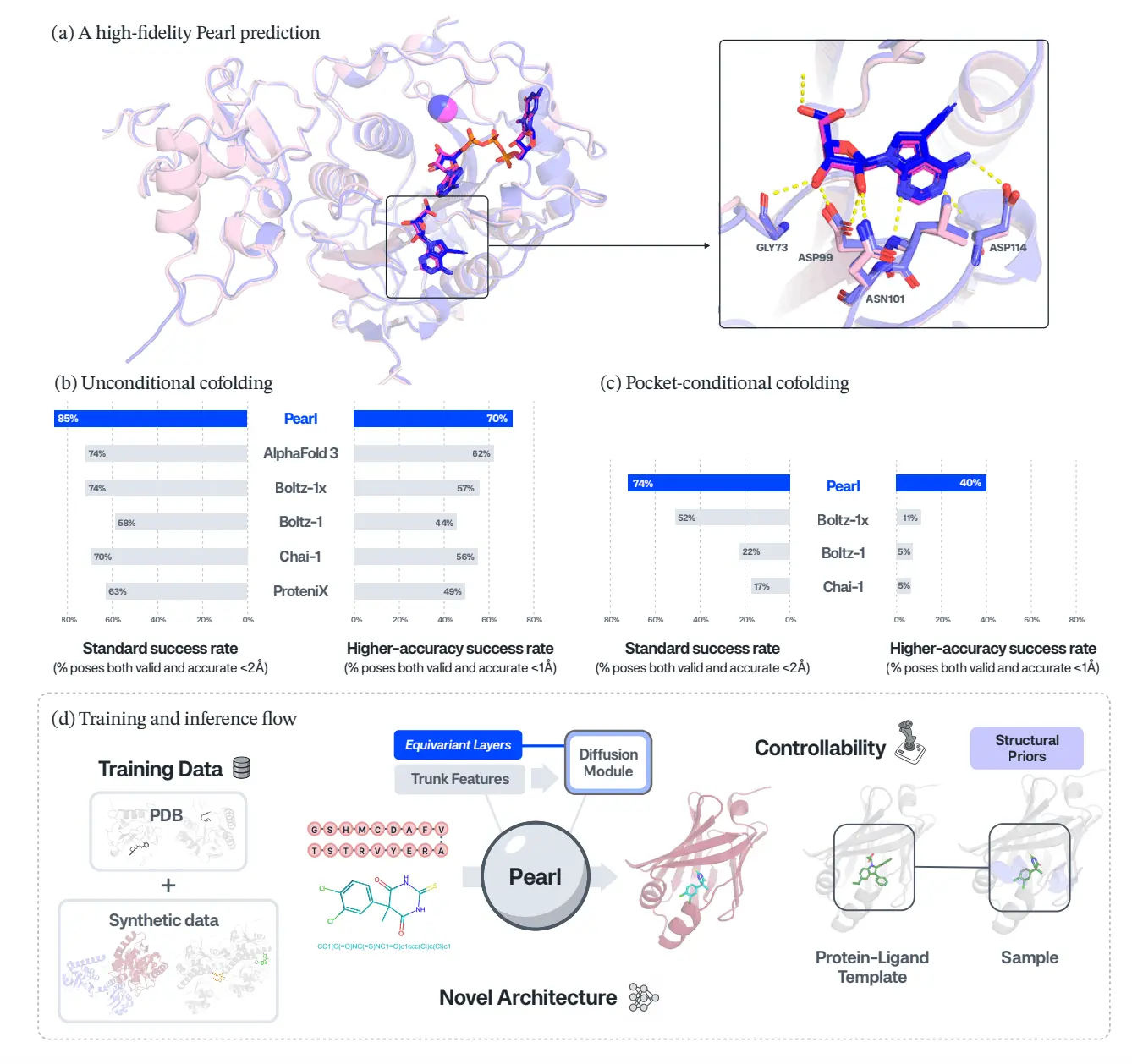
For the headline benchmarks, Pearl not only outperformed the competitors in the field, but it also significantly surpassed the field. In Runs N’ Poses, Pearl has an 85.2% success rate and is not only valid in the pose(ligand RMSD < 2 Å), but is also valid in the physical structures, which is a 14.5% relative performance increase over AlphaFold 3 and Boltz-1x within the same protocol. On PoseBusters, Pearl has a success rate of 84.7%, which is a 14.2% relative increase over the next best performing open model and is nearly a 40% increase over the official reported success rate by AlphaFold 3. What is most important is that the success rate does not drop significantly when the more stringent requirements of physical plausibility on the structures are applied. This proves that the poses generated by Pearl were not only very close in RMSD but also very close in their chemical validity.
High-Accuracy Poses for Real Medicinal Chemistry
The authors push beyond the usual RMSD < 2 Å metric and focus on the more demanding RMSD < 1 Å. They also added the PoseBuster validity requirement, which is considered the more stringent requirement, and is a more practical requirement needed for medicinal chemists for the SAR and potency modeling. Pearl is the only model to maintain over 70% success at this tight requirement for the high accuracy threshold. In contrast, other models performing this task fall short of the requirement by a wide margin. On Genesis’ proprietary InternalXtals dataset—111 challenging drug-program structures—Pearl delivers a nearly four-fold relative gain over the strongest open baseline at RMSD < 1 Å, even though that baseline has access to more recent training data.
Two Modes: Discovery and Optimization
Pearl aims to replicate the workflows of drug discovery teams as closely as possible. In unconditional mode, it only uses the protein sequences and 2D ligand topologies (and MSAs plus templates) to explore binding poses. This is best for early exploration when the pockets are not well understood. In “pocket-conditional” mode, Pearl takes as guidance an apo or holo structure and a small set of pocket residues, allowing chemists to constrain the pose prediction to specific known binding sites and cofactors. Pearl is still the leader in this conditional mode as well, beating the competition in PoseBusters and InternalXtals, and making the most gains at the 1 Å threshold, which is of vital importance for lead optimization.
Synthetic Data and Scaling Laws in Molecular AI
The synthetic data the authors discuss is the most fascinating. Pearl is trained not only on curated PDB structures and distilled monomer data, but also on a large set of protein-ligand complexes. Pearl also uses public structures for building synthetic data. Special experiments with smaller nightingales showed a clean scaling curve as the fraction of synthetic complexes during training increased. This is presented as early evidence of “synthetic data scaling laws” in drug discovery, suggesting that high-quality simulation can play a role for molecular AI similar to simulation in autonomous driving.
Architecture, Hardware, and Practical Throughput
Under the hood, Pearl’s trunk uses efficient triangle operations, accelerated with NVIDIA’s cuEquivariance kernels, and a conservative mixed-precision strategy that pushes heavy computation into bfloat16 while keeping numerically sensitive parts in full precision. This yields about a 22% training speedup from mixed precision alone and an additional ~15% speedup in training, plus 10–80% faster inference compared with a vanilla PyTorch baseline, depending on sequence length. Those gains are not academic: they are what make it feasible to train on large synthetic corpora and deploy Pearl at an industrial scale inside Genesis’ GEMS platform.
A Foundation for the Next Era of Molecular AI
The authors are candid that Pearl is not magic. Within case studies, authors find instances where ligands have been mapped to areas where they don’t belong across training pockets. There are also instances where loop rearrangements are overfitted, or a completed structure visual exhibits an incorrect reference distance and/or is missing key hydrogen atoms.
Still taken together, according to the Pearl Technical report, Genesis Molecular AI is becoming one of the front-running companies in the development of operationally useful, scientifically grounded foundational models. Pearl successfully transitioned from just impressive demos to genuine utility in the hands of medicinal chemists. This was accomplished through state-of-the-art synthetic data generation, sophisticated hardware-aware engineering, and the groundbreaking entry of an equivariant generative modeling technique into the field. For innovators in the field of AI and drug discovery, the takeaway is simple: the Data generation coupled with physical inductive biases and controllable inference will be the three mainstays of the molecular models of the future.
Article Source: Reference Paper | Reference Article
Disclaimer:
The research discussed in this article was conducted and published by the authors of the referenced paper. CBIRT has no involvement in the research itself. This article is intended solely to raise awareness about recent developments and does not claim authorship or endorsement of the research.
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Follow Us!
Learn More:




Anchal is a consulting scientific writing intern at CBIRT with a passion for bioinformatics and its miracles. She is pursuing an MTech in Bioinformatics from Delhi Technological University, Delhi. Through engaging prose, she invites readers to explore the captivating world of bioinformatics, showcasing its groundbreaking contributions to understanding the mysteries of life. Besides science, she enjoys reading and painting.











{kind=link}