
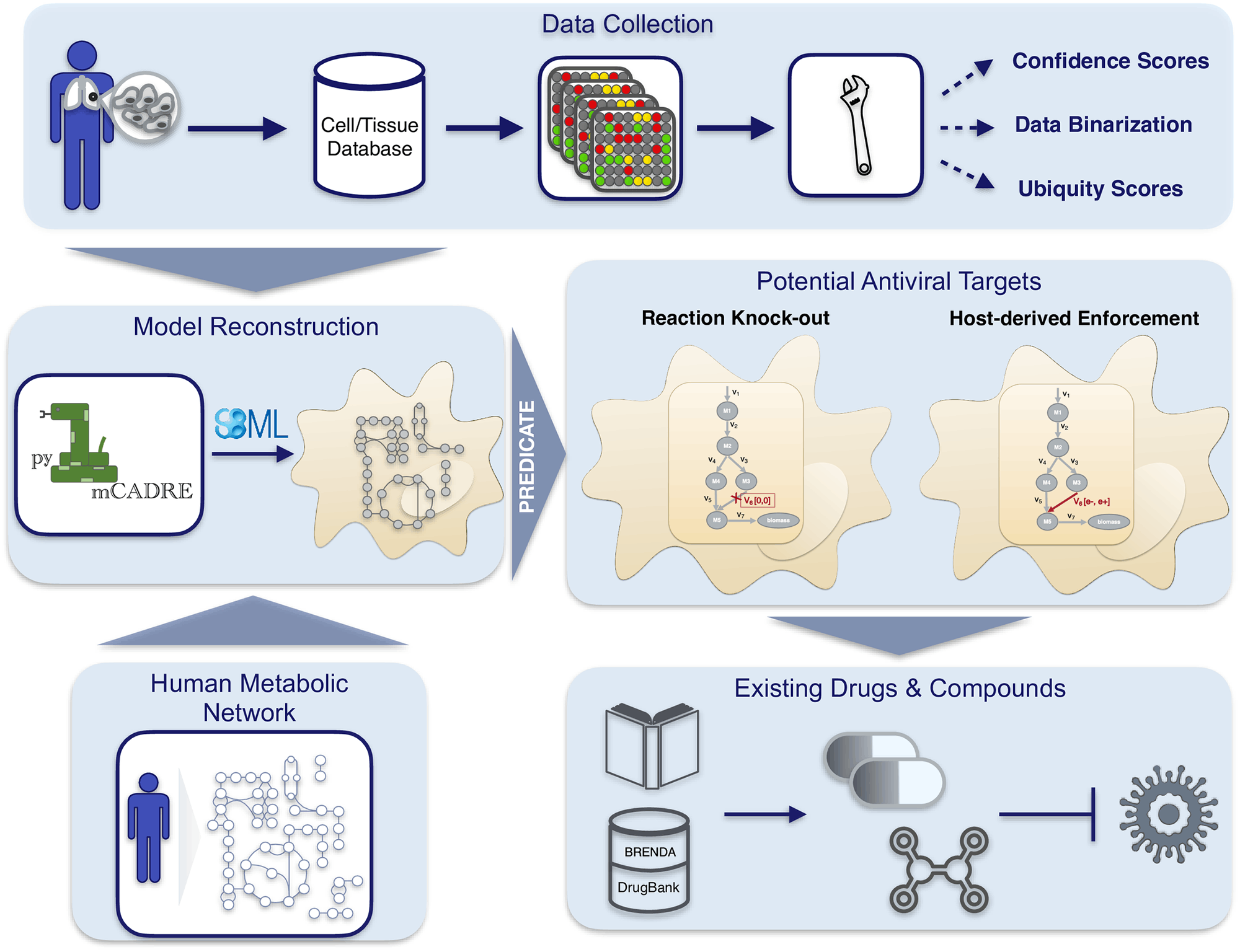
Scientists from the University of Tübingen, Germany, have developed a novel workflow for predicting robust druggable targets against emerging viral infections such as the SARS-CoV-2. The workflow implementation comprises pymCADRE and PREDICATE, which incorporate metabolic changes in infected cells for accelerated prediction of drug targets. The authors generated tissue-specific metabolic models, constructed viral biomass functions, and predicted host-based antiviral targets from several genomes. The accelerated workflow was applied to create a metabolic network of primary bronchial epithelial cells that were infected with SARS-CoV-2. This identified and predicted enzymatic reactions with inhibitory effects. Highly promising targets were predicted by the workflow from the purine metabolism networks and also identified the carbohydrate and pyrimidine metabolism networks to enhance viral inhibition.
Why do we need a novel workflow for predicting antiviral targets?
Viral infections have been a cause of concern among the medical as well as the scientific community. The 2019 pandemic driven by the SARS-CoV-2 virus has further emphasized the need for bioinformatics tools and a system biology approach for identifying suitable drug targets for antivirals. While vaccines against viral infections, especially during the SARS-CoV-2 pandemic, have proved significant in reducing mortality amongst the cohort of patients with co-morbidities, the effect of vaccines is shortlived and calls for therapeutic measures such as drug development against the viral infection.
Antiviral drug development greatly involves engineering the host metabolism. This is largely due to the fact that viruses replicate within the host cells and use up the host cell’s energy for the mass production of viruses via replication and reprogramming of the host cell genome. One of the largest classes of small-molecule antiviral drugs targets metabolic enzymes involved in nucleotide synthesis. Several studies involving bacterial infections have also confirmed that host cell metabolism engineering can regulate viral growth. Thus, the impact of viral biosynthesis on host metabolism and the effects of engineered host metabolism on viral growth reduction is of paramount importance in drug development and prediction of druggable targets against viral infections.
While viruses involved in viral infections typically evolve and acquire several mutations pertaining to errors in copying mistakes during replication which often are druggable targets against the disease-causing pathogen. However, in the case of SARS-CoV-2, the number of such mutations is quite low owing to the presence of error-correcting and proofreading enzymes being present, thus drastically reducing the options for therapeutic intervention. Thus, a system biology-based approach involving the reconstruction of cell-specific Genome-scale Metabolic Models (GEMs) recapitulating the metabolism of particular cell types is the need of the hour.
Previous efforts in identifying drug targets against SARS-Cov-2 have applied constraint-based metabolic modeling involving a vast plethora of resources and approaches. A recent study uses a draft model of the airway epithelial cells from Recon1 and, after refining it using Recon3D, predicts druggable targets against covid-19. However, the use of pre-existing reconstruction tools and models for obtaining the tissue metabolism representation is not ideal. Thus, the need for a novel and accelerated workflow for the prediction of druggable targets against viral infections using metabolic changes in the host cells.
pymCADRE: Python implementation of metabolic Context-specificity Assessed by Deterministic Reaction Evaluation
In 2012, mCADRE was developed to construct metabolic models using human gene expression data and network topology information. However, this was implemented in MATLAB and thus not freely available to the scientific community. It is also based on the first version of the human model, RECON1. Thus the need for novel workflow development.
pymCADRE is a re-implementation of mCADRE in python and incorporates scripts for data processing, enabling a better and faster generation of metabolic models. Using the available metabolic network in RECON1, the authors generated a novel tissue-specific model of primary Human Bronchial Epithelial Cells (HBECs). The authors next simulate SARS-CoV-2 infection of these epithelial cells using the viral structural information.
PREDICATE: Prediction of antiviral targets
The immediate next step in the workflow involved developing a fully automated computational tool for the prediction of antiviral targets. The authors implemented a python tool using the stoichiometric approaches used by Aller et al. and constructing a single Viral Biomss Objective Function (VBOF). This generates an integrated host-virus model, which the authors use to predict cellular metabolic pathways whose inhibition can lead to the suppression of virus replication. Hence, potentially druggable targets are predicted against the viral disease.
The following figure illustrates the workflow:
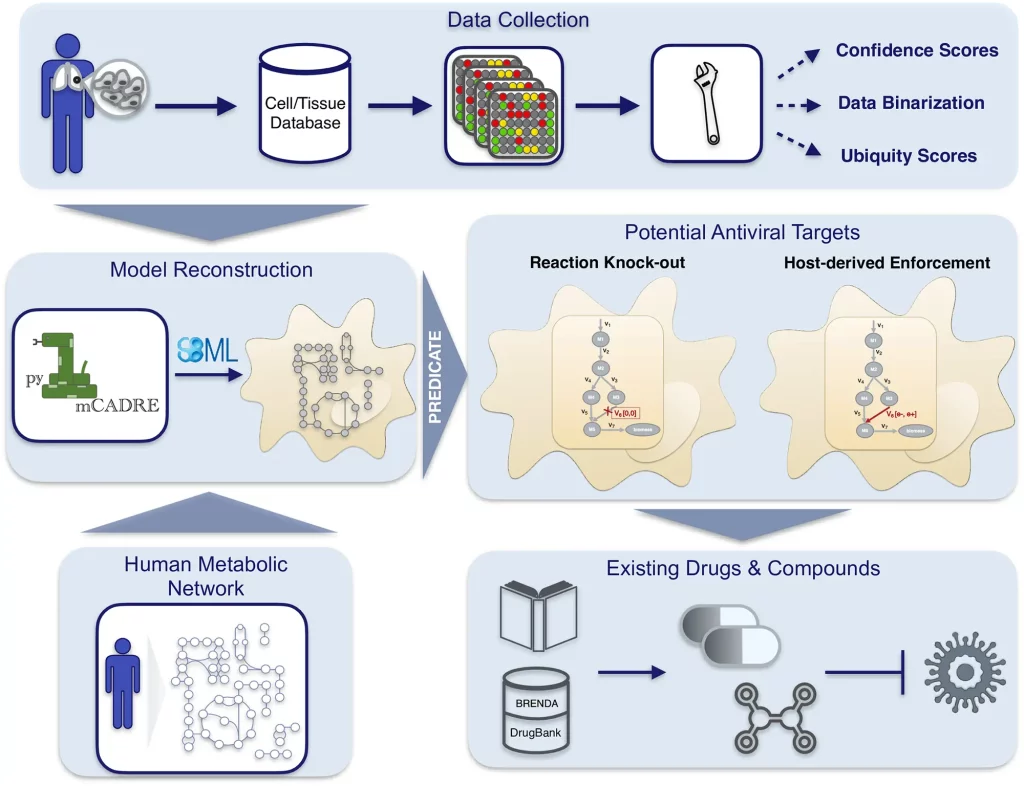
Image source: https://doi.org/10.1371/journal.pcbi.1010903
Performance and results
pymCADRE was found to proceed faster than mCADRE when Flux Variability analysis was performed. These two models rendered 100 % overlap for reactions.
The simulated infection model recovered already known antiviral drug targets for Chikungunya, Dengue, and Zika viruses, thereby establishing the robustness of the approach. The simulated infection model also verified GK1 as a target for suppressing the SAR-CoV-2growth without harming the host, thus a potential drug target. The method also confirmed previously mentioned glycolytic targets from the literature. The method predicted novel drug targets against SARS-CoV-2, CTP synthase 1 (CTPS1), which greatly reduced the viral growth as by GK1. The authors also show that predicted targets are robust against all known variants of concern of the SARS-CoV-2. They illustrate the possibility of using existing drugs and inhibitors against predicted enzymes to suppress the growth of the virus.
Conclusion
The authors developed an automated computational tool for predicting druggable targets against viral infection using the information on the host cell’s metabolic changes. The workflow involves two components involving the construction of tissue-specific metabolic models and the prediction of druggable targets. The tools are seen to perform with high efficacy and robustness and identify novel drug targets for the SARS-CoV-2 virus. The authors envision future iterations of the tool to incorporate complex models such as in RECON2. Needless to say, this bioinformatic and system biology-based approach is a remarkable feat for drug development and therapeutics development against viral infections and SARS-CoV-2, in particular.
Article Source: Reference Paper | Reference Article
Learn More:




Banhita is a consulting scientific writing intern at CBIRT. She's a mathematician turned bioinformatician. She has gained valuable experience in this field of bioinformatics while working at esteemed institutions like KTH, Sweden, and NCBS, Bangalore. Banhita holds a Master's degree in Mathematics from the prestigious IIT Madras, as well as the University of Western Ontario in Canada. She's is deeply passionate about scientific writing, making her an invaluable asset to any research team.





{kind=link}