
Scientists from the Max Planck Institute of Molecular Physiology, Germany, have developed a method, TomoTwin, an open-source general picking model for cryogenic-electron tomograms. The method is based on deep metric learning and incorporates the embedding of tomograms in an information-rich, high-dimensional space that separates macromolecules according to their three-dimensional structures. The localization of particles within the tomogram volume is a crucial first step for a detailed analysis of macromolecules through subtomogram averaging. However, issues like low signal-to-noise ratio and cellular area crowding make this task difficult. Currently, available methods are either error-prone or necessitate manual annotation of training data. TomoTwin enables users to identify proteins in tomograms anew without requiring manual annotation of training data or retraining of the network for locating new proteins.
Why do we need TomoTwin?
Cryogenic-electron tomography (Cryo-ET) is a cutting-edge technology for seeing macromolecules in their natural cellular environments. Cryo-ET captures cellular processes in three dimensions and great detail. Subsequent analysis of specific macromolecules from tomograms through subtomogram averaging (STA) enables in situ macromolecular complex’s structural determination. In fact, STA acts as a powerful bridge between cellular proteomics and protein biochemistry. However, a crucial requirement for successful STA is the localization of macromolecules within the tomogram, which is often met with limitations pertaining to the three-dimensional nature of the data.
Deep learning-based approaches developed by Moebel et al. and Hao et al. address the localization problem for studying cellular processes at the mesoscopic scale. These typically incorporate 3D-Unet convolutional neural network (CNN)) architectures. However, generalization in terms of proteins has not been realized until now. For each protein of interest, these methods require manual annotation of thousands of particles in tomograms as well as training of the neural network for identifying that protein.
The authors developed TomoTwin to address these limitations using the deep metric learning paradigm.
Deep metric learning: The philosophy behind TomoTwin
The deep metric learning paradigm involves encoding data into high-dimensional representations called embeddings. In the embedding space, the model is rewarded for placing data from the same class in close proximity and penalized for placing data from different classes nearby during training. Over the course of the training process, the model learns to cluster similar classes into a distinct region in the embedding space and place dissimilar ones further apart. Such ordering of embeddings often enables de novo identification of classes in the embedding space. Thus, such models based on the deep metric learning approach have presented the possibility of generalization in terms of placing new classes of data into the embedding space based on their similarity to known classes without retraining. This is exactly what was needed for Cryo-ET.
The authors thus developed TomoTwin, a novel deep metric learning toolkit for generalized particle picking in cryo-electron tomograms. The toolkit presents two workflows for macromolecular localization: a reference-based workflow for picking a single molecule for each protein of interest and a novel clustering workflow for identifying macromolecular structures of interest on a 2D manifold.
TomoTwin workflow: a brief overview
The two workflows mentioned above together form the TomoTwin workflow:
- The first step involves embedding the tomogram with the pre-trained model.
- References can also be selected and embedded to create target embeddings.
- The clustering workflow involves projecting the tomogram embeddings onto a 2D manifold and the selection of clusters of interest to generate target embeddings using an interactive lasso tool.
- The distance matrix between target embeddings and embeddings of the tomogram is calculated.
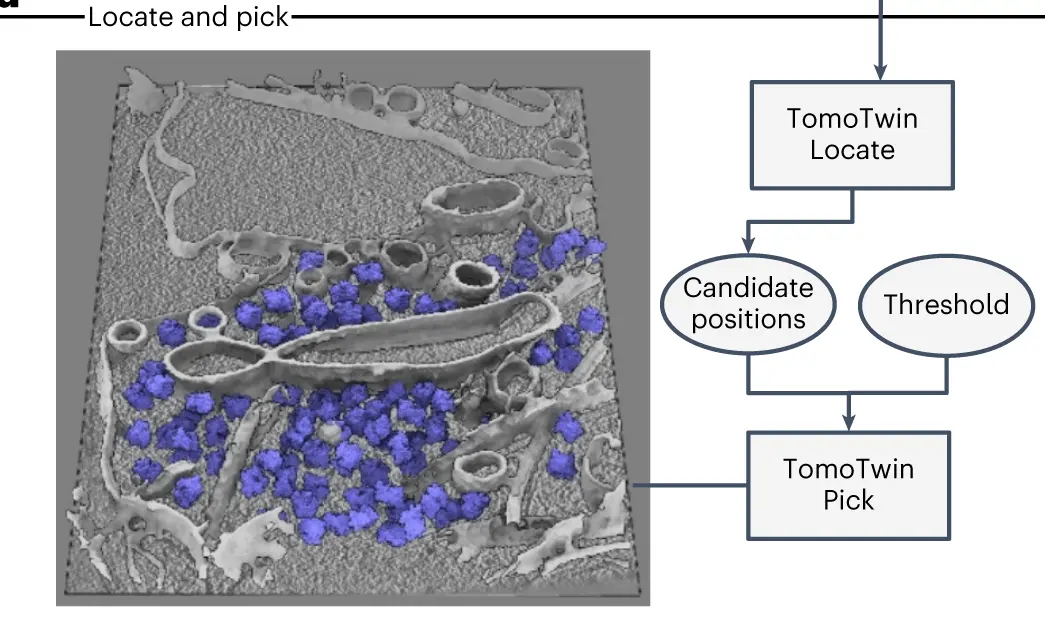
- Local maxima are all located using TomoTwin Locate. These maxima are then used to pick the final coordinates for each protein of interest using TomoTwin Pick. This involves size and confidence thresholding.
Performance of TomoTwin
TomoTwin has been shown to successfully generalize the picking of particles in Cryo-ET.
- The picking model has been shown to generalize across protein shape and size.
- It has also been shown to generalize to unseen proteins.
- TomoTwin picks proteins accurately in experimental tomograms and generalizes them across a variety of experimental setups.
- TomoTwin has been shown to be highly accurate in particle picking as compared to other particle-picking methods available.
Conclusion
Particle picking is a crucial step in Cryo-ET, and previous methods have faced several challenges in generalizing the methodology of particle picking across proteins of interest. TomoTwin, based on the deep metric learning paradigm, remarkably does away with the limitations posed by the earlier methods. It is a robust, open-source toolkit for particle localization in cryo-electron tomograms using deep learning techniques with very high accuracy. The code for TomoTwin training and development, as well as for the TomoTwin general picking model, are freely available. This will be a reliable toolkit for several Cryo-ET-based experiments, further our knowledge of protein dynamics, and aid in drug delivery.
Article Source: Reference Paper | TomoTwin: GitHub Link
Learn More:




Banhita is a consulting scientific writing intern at CBIRT. She's a mathematician turned bioinformatician. She has gained valuable experience in this field of bioinformatics while working at esteemed institutions like KTH, Sweden, and NCBS, Bangalore. Banhita holds a Master's degree in Mathematics from the prestigious IIT Madras, as well as the University of Western Ontario in Canada. She's is deeply passionate about scientific writing, making her an invaluable asset to any research team.





{kind=link}