
The multi-adaptive Support Vector Machine Learning Algorithm (maSVM) can improve comparisons between binary Protein-Protein interaction datasets gathered in different laboratories and can be utilized to confidently prioritize potential targets for Protein-Protein Interaction (PPI) based drug discovery. The researchers first showed that the automatic classification of large quantitative interaction datasets could be performed with high confidence. The study shows that combining high-quality quantitative binary interaction data, AI-based scoring systems, and computational modeling with wet lab methodologies can help prioritize PPI targets for the development of novel therapeutics.
Protein-Protein Interaction as Drug Target
Proteins and their interacting partners are one of the most common and preferential targets in Drug lead discovery and screening. So, targeting enzymes, Ion channels, and Receptors to block the cross-talk that provokes disease progression can potentially serve this purpose. But often, the identification and validation of Protein-Protein interactions experimentally is a relentless, time-consuming, and thus unfeasible approach.
Here, the bioinformatics tool comes to the rescue. For example, in-silico tools that generate the structure of a protein, compute its interaction partners, perform docking, and screen lead compounds are generally implemented in such endeavors. Artificial Intelligence-based tools, AlphaFold and RoseTTAFold, are the most advanced, accurate, and widely utilized to generate protein’s 3D structural models, simulate Protein assembly, and analyze interface surfaces. The VirtualFlow platform can be used to screen billions of compounds in silico against the predicted target. Complementing experimental assays with Computer-aided tools offers more reliability and specificity and is cost & time effective.
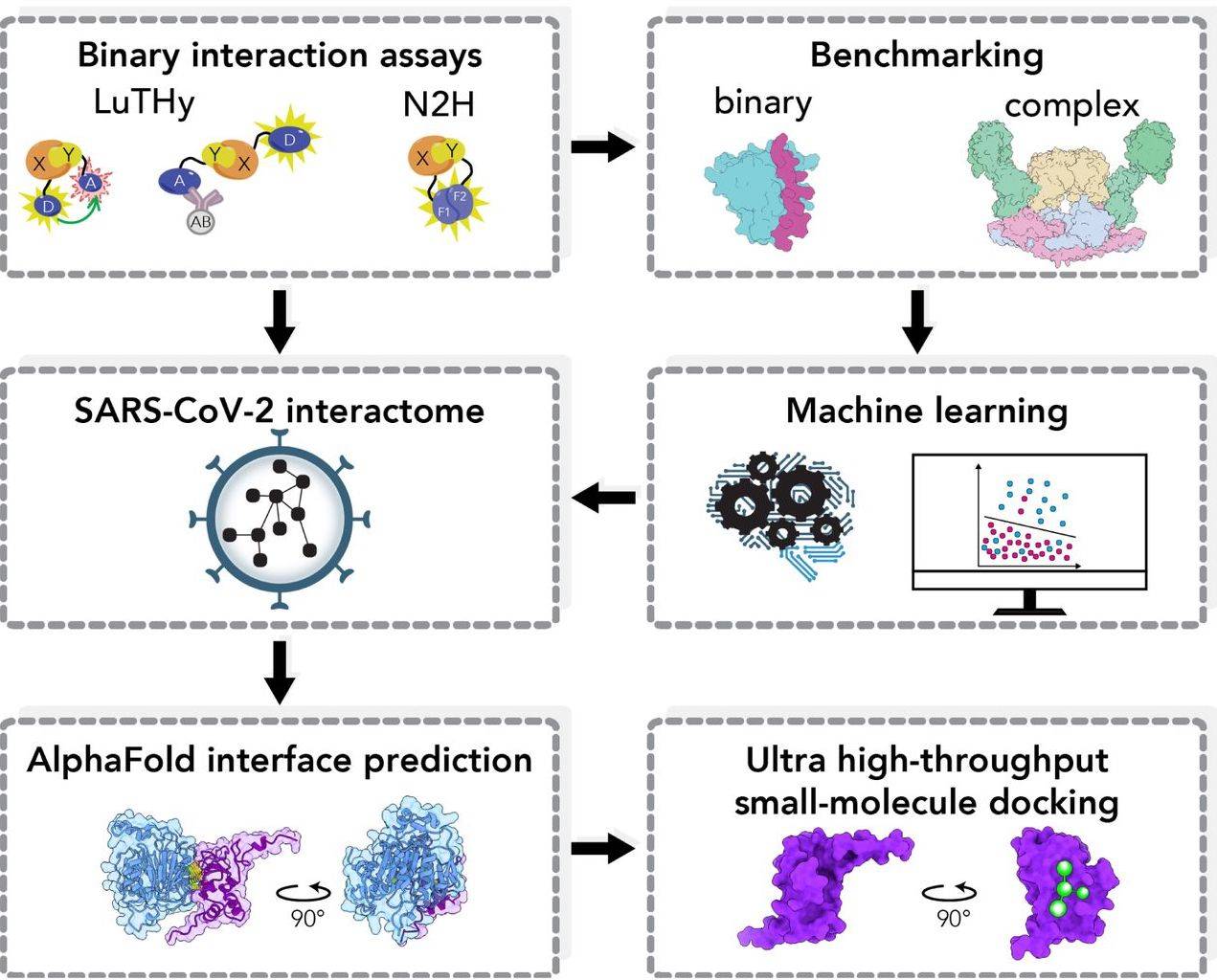
The Pipeline Overview
The pipeline integrates multiple techniques to streamline the process of PPI-based drug discovery. The researchers have developed a machine-learning approach that leverages quantitative data from binary PPI assays and AlphaFold-Multimer predictions to prioritize interactions. By employing the LuTHy assay in conjunction with a machine learning algorithm, researchers successfully identified high-confidence interactions among SARS-CoV-2 proteins and determined their three-dimensional structures using AlphaFold Multimer.
Targeting the NSP10-NSP16 SARS-CoV-2 methyltransferase complex, the researchers utilized VirtualFlow for ultra-large virtual drug screening to pinpoint the contact interface. Through this screening, researchers identified a compound capable of binding to NSP10, inhibiting its interaction with NSP16 and disrupting the methyltransferase activity of the complex, thereby impeding SARS-CoV-2 replication.
Scoring Protein-Protein interaction (PPI) data gathered from different binary PPI assays to shortlist high-confidence interacting partners can often be erroneous and non-reproducible due to the lack of coherent and robust approaches. The maSVM algorithm implemented in the study demonstrated its advantage in scoring the Protein-Protein Interaction data gathered from different assays as it displayed reproducibility, specificity or sensitivity, and robustness. Moreover, the algorithm can be applied to systematically determine coupled interactions within a multi-protein complex.
Advantages of PPI Targeting
Traditional drug discovery efforts have primarily focused on enzymes, ion channels, and receptors. However, the number of newly approved drugs has decreased over the years, indicating a need for exploring alternative therapeutic targets. PPIs offer a promising avenue, as they are crucial in signaling pathways and multisubunit complexes. Expanding the druggable proteome by targeting PPIs can unlock new classes of drug targets and potentially overcome immune evasive properties caused by pathogen mutations.
Integration of Experimental and Computational Approaches
To comprehensively explore PPIs, experimental techniques such as affinity purification coupled to mass spectrometry (AP-MS) and binary PPI assays like yeast two-hybrid (Y2H) with structural biology methods such as cryo-electron microscopy (cryo-EM) were combined. These techniques enable the identification, characterization, and visualization of protein complexes and their interaction interfaces. However, due to the limited number of experimentally resolved protein complex structures, computational predictions have emerged as valuable tools.
Recent advances in AI-based structure prediction algorithms, such as AlphaFold and RoseTTAFold, have significantly improved the accuracy of protein complex modeling and interaction interface prediction. The researchers show how these predictions, coupled with experimental validation, provide a robust foundation for identifying and targeting PPIs.
Additionally, the researchers highlight the utility of VirtualFlow, an in silico method capable of screening billions of compounds against predicted targets. This approach offers a time- and cost-effective means to identify potential PPI inhibitors. The research work presents the successful application of VirtualFlow to target the NSP10-NSP16 interaction and the subsequent identification of a compound with inhibitory effects on SARS-CoV-2 replication.
Conclusion
In this study, the researchers presented a comprehensive pipeline to bridge the gap between experimental and computational approaches and accelerate the identification and validation of PPI targets. The machine learning approach analyzes the Protein interaction data. The maSVM algorithm can reduce the variability between recovered interactions in different experiments and can compute probabilities for being a true interaction of the protein pair tested. They demonstrated that the maSVM algorithm is universally applicable to classify binary PPIs from different quantitative datasets, including the interface analysis of AlphaFold predicted complex structures. Combining such in silico and laboratory procedures for validation of Protein-Protein interactions as well as for drug screening would speed up the process of developing therapeutics.
Article Source: Reference Paper
Important Note: bioRxiv releases preprints that have not yet undergone peer review. As a result, it is important to note that these papers should not be considered conclusive evidence, nor should they be used to direct clinical practice or influence health-related behavior. It is also important to understand that the information presented in these papers is not yet considered established or confirmed.
Learn More:




Aditi is a consulting scientific writing intern at CBIRT, specializing in explaining interdisciplinary and intricate topics. As a student pursuing an Integrated PG in Biotechnology, she is driven by a deep passion for experiencing multidisciplinary research fields. Aditi is particularly fond of the dynamism, potential, and integrative facets of her major. Through her articles, she aspires to decipher and articulate current studies and innovations in the Bioinformatics domain, aiming to captivate the minds and hearts of readers with her insightful perspectives.











{kind=link}