
Scientists from CHOP, Philadelphia, and UCLA have developed an integrated computational workflow to discover novel cancer immunotherapy targets from pre-mRNA alternative splicing (AS). The prevalence of alternate splicing across different cancers is wide, yet the vast repertoire of cancer immunotherapy targets remains unexplored. IRIS has been developed to identify AS-derived tumor antigens (TAs) for T cell receptor and chimeric antigen receptor T cell therapies. IRIS incorporates multi-screening data and leverages large-scale tumor and normal transcriptome data. The author performed a proof-of-concept analysis that integrates transcriptomics and immunopeptidomics data to show that hundreds of IRIS-predicted TCR targets are presented by human leukocyte antigen (HLA) molecules. They also applied the workflow for the target discovery of neuroendocrine prostate cancer.
Alternative splicing and cancer cell proteome
Post-transcriptional RNA processing is crucial to generating the eukaryotic proteome. The dysregulation of it has a significant impact on the cancer cell proteome. Several such processes lead to the production of aberrant proteins as well as aberrant immunogenic peptides. Studies have shown that tumors harbor around 30% more alternative splicing events than normal tissues. Cancer immunotherapy has seen remarkable progress in recent years. Antibodies like PD-1 and CTLA-4 reactivate tumor-specific T-cells and are seen to be quite effective. The insightful discovery that cancer cells express specific T-cell antigens has led to a major effort toward antigen discovery. However, the discovery of tumor antigens (TAs) remains elusive. Although somatic mutation-derived TAs have been successfully targeted by cancer therapies, such a methodology is not very effective for tumors with a low or moderate mutation load.
Available tools for discovering AS-derived TAs
Currently, available tools for AS-derived TA discovery are scanty. Two computational tools, ASNEO and NeoSplice, have been developed for the discovery of AS-derived TCR targets for cancer immunotherapy. These tools are based on using RNA-seq data from tumors as well as normal tissues to identify putative tumor-specific AS events. This is followed by the HLA binding prediction. However, these tools lack the computational infrastructure to tackle large cohorts of tumor and transcriptomic data for comprehensively determining the tumor association and specificity of predicted targets.
IRIS: Isoform peptides from RNA splicing for immunotherapy target screening
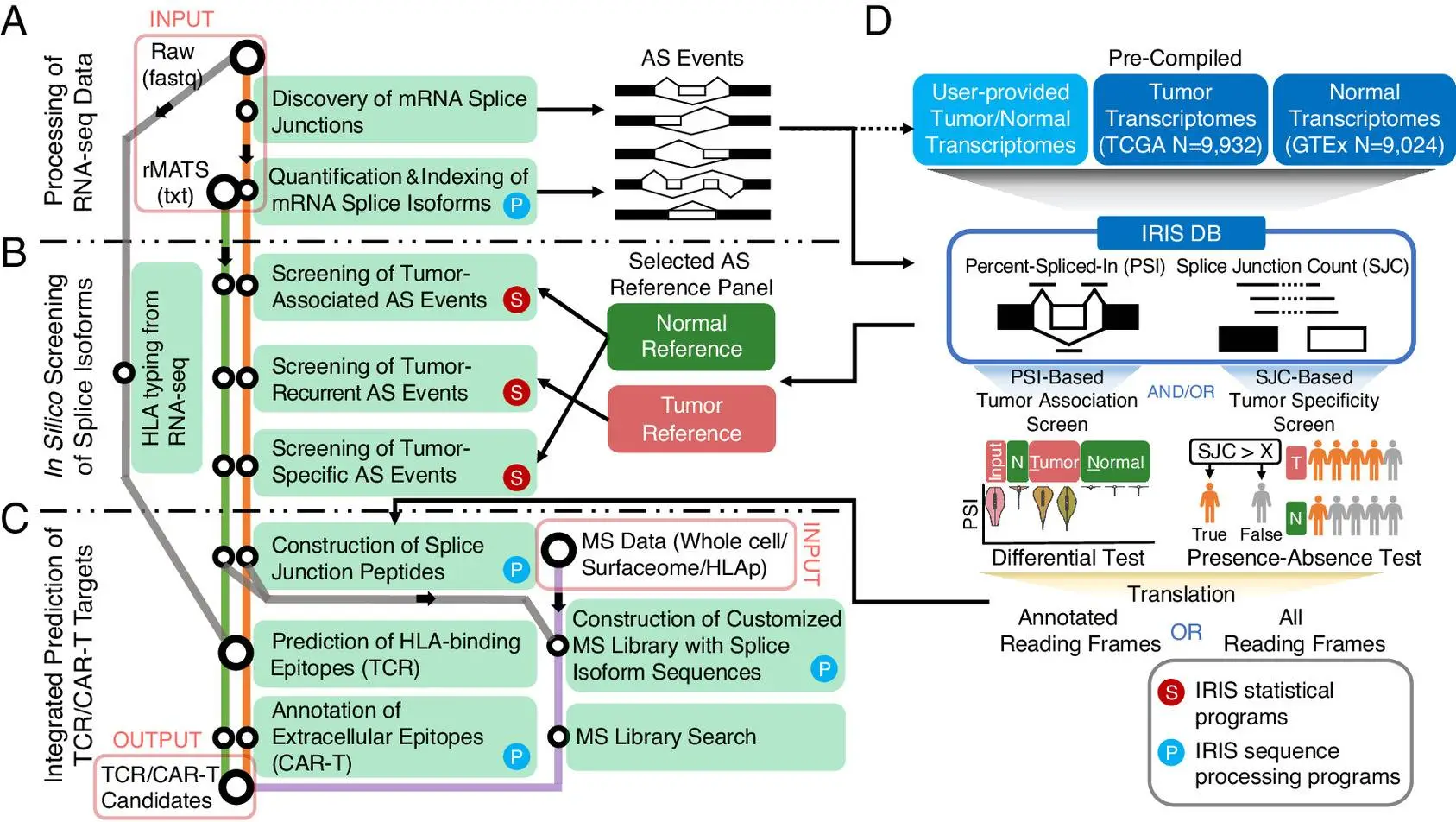
The authors developed an in silico platform for identifying and characterizing AS-derived immunotherapy targets of varying degrees of tumor association as well as specificity. This is achieved by using an AS reference representing the splicing profiles of tens of thousands of tumors as well as normal transcriptomes generated by large-scale consortia such as GTEx and TCGA. The computational workflow is data-informed and powered by the software rMATS-turbo for analyzing RNA-seq data for AS events in an ultrafast manner and with improved efficacy.
The computational workflow of IRIS has three main computational modules: RNA-seq data processing, in-slico screening, and TCR/CAR-T target prediction. A reference database of AS profiles across tumors and normal tissue is also included. The workflow also includes two screening tests to assess tumor association and specificity.
The following figure illustrates the workflow of IRIS:
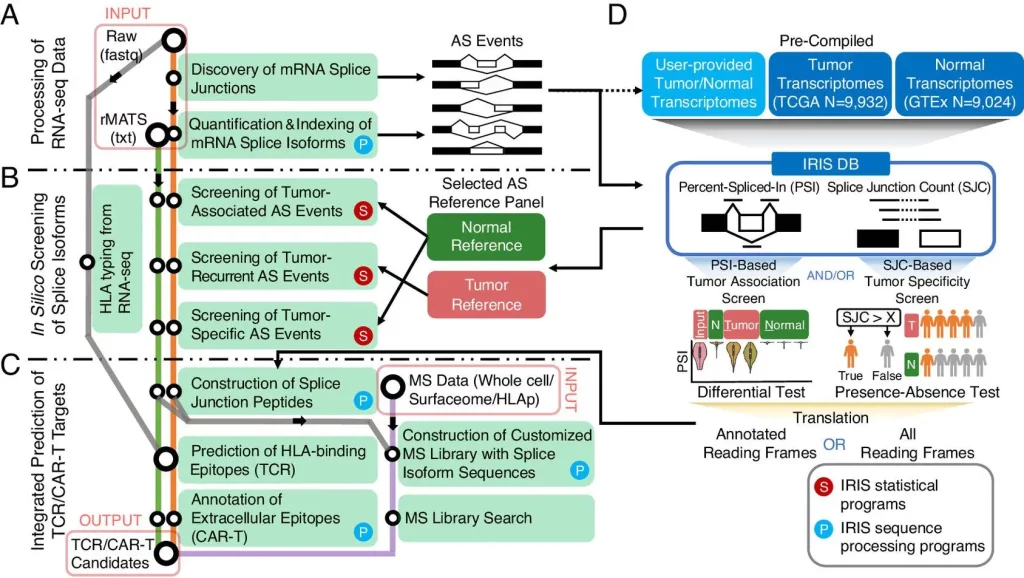
Image source: https://www.pnas.org/doi/10.1073/pnas.2221116120
Proof-of-concept analysis
In this analysis, the authors set out to discover and characterize AS-derived peptides that are presented by HLA molecules. The authors analyzed two B lymphoblastoid cell lines using their paired RNA-seq data and immunopeptidomics data, as well as that of a cancer cell line. This analysis established that AS-derived peptides are present in cell line immunopeptidomes.
Immunotherapy target discovery
The authors applied the IRIS workflow to the RNA-seq dataset of 23 NEPC samples. IRIS predicted immunotherapy targets for these NEPC samples. IRIS also discovered that skipped exon (SE) events are enriched for microexons. IRIS also enables the visualization of AS-derived immunotherapy targets in the form of an integrated report that allows users to visualize the predicted targets based on multiple criteria. IRIS further characterized TCRs reactive to IRIS-predicted NEPC epitopes.
Conclusion
While immunotherapy has revolutionized cancer therapeutics, the repertoire of antigens, including that for several pediatric cancers, is incomplete. The authors addressed this scarcity and developed the IRIS toolkit for discovering novel immunotherapy targets of cancers using RNA AS events. The increased computational speed compared to other available tools as well as the capability of IRIS to handle big data, makes it a remarkable and groundbreaking discovery in terms of tool development for the scientific as well as the therapeutic community. With more targets being identified, several cancers could be addressed readily and effectively, thereby changing the lives of millions and global health worldwide. The potential of therapy development with the new set of novel targets will indeed revolutionize immunotherapy and cancer treatment.
Article Source: Reference Paper | Article Reference | IRIS: Software
Learn More:




Banhita is a consulting scientific writing intern at CBIRT. She's a mathematician turned bioinformatician. She has gained valuable experience in this field of bioinformatics while working at esteemed institutions like KTH, Sweden, and NCBS, Bangalore. Banhita holds a Master's degree in Mathematics from the prestigious IIT Madras, as well as the University of Western Ontario in Canada. She's is deeply passionate about scientific writing, making her an invaluable asset to any research team.





{kind=link}