
TransFlow (Transmission Workflow), a convenient, quick, effective, and thorough WGS-based transmission analysis pipeline, is presented by Chinese researchers. TransFlow integrates a number of cutting-edge technologies to perform transmission analysis on raw sequencing data. This analysis includes quality control, sequence alignment, variant calling, downstream transmission grouping, transmission network reconstruction, and transmission risk factor inference. TransFlow is easily adaptable to any computing environment and depends on Snakemake and Conda to determine relationships between subsequent processing stages.
Tuberculosis (TB), which is caused by the bacteria Mycobacterium tuberculosis complex (MTBC), continues to pose a serious danger to worldwide public health because it results in significant mortality brought on by a single infection. Mycobacterial interspersed repetitive-unit-variable-number tandem repeat (MIRU-VNTR) methods, and sequence-based genotyping assays (multi-locus sequence typing, MLST) have been replaced in recent years by next-generation sequencing (NGS) based whole-genome sequencing (WGS) for the molecular detection of TB outbreaks.
A growing number of studies are using whole-genome sequencing (WGS) to better understand how Mycobacterium tuberculosis (MTB) spreads. Using the WGS approach, the epidemiological investigation of TB necessitates the use of a wide range of bioinformatics tools. It might be challenging to use these analysis tools effectively in a configurable and replicable fashion, especially for novices.
Genomic surveillance provides for the sensitive detection of epidemics and the analysis of the spread of illness throughout the population by integrating molecular data with traditional epidemiological data.
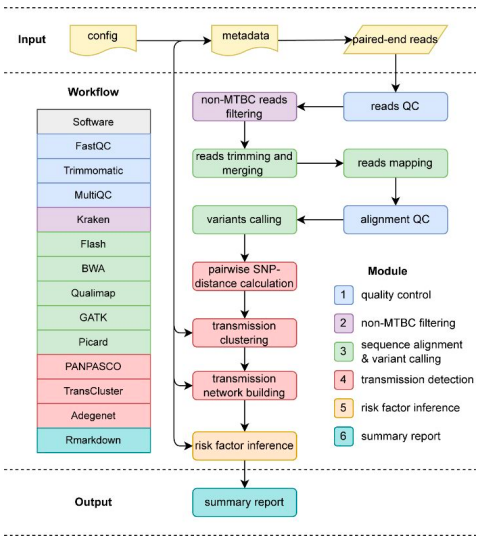
(Transmission Workflow). The different modules of the pipeline are broken down by
colors.
Image Source: https://doi.org/10.1093/bioinformatics/btac785
A unique workflow called TransFlow combines many of the cutting-edge techniques presently used in WGS-based MTBC transmission research into a single, quick, and user-friendly pipeline using a contemporary computational workflow management system, Snakemake. TransFlow is scalable because it can be used on both computing servers with plenty of cores (which enable parallel computation) and personal computers with little in the way of processing power. TransFlow is also adaptable and customizable; depending on the user’s settings and inputs, it may further combine additional epidemiological data for molecular surveillance and adopt both SNP-based and transmission-based methodologies for transmission clustering. To demonstrate the workflow’s capabilities and performance, two actual WGS datasets were used from the CTB and UKTB projects. To speed up the assessment and implementation of workflow, drafting documentation, example data, outputs, and a prototype report available on the official GitHub repository is necessary.
Three fundamental ideas permeate every aspect of the pipeline’s architecture in TransFlow. To begin with, it is created with the visualization of findings as a fundamental design element to provide output that captures significant analytical results in educational, publication-quality figures.
Furthermore, TransFlow was created using Snakemake in order to be both efficient and customizable. Last but not least, researchers wanted to make sure that anybody, including those with no background in bioinformatics, may install and utilize TransFlow. As a result, installing TransFlow needs little user involvement, and the procedure may be started with only one terminal command and inputs from any text or table editor.
TransFlow’s analysis phases are described in terms of “rules” that link input files to output files as part of the workflow as a whole. Snakemake executes and deduces the set of rules required to produce a “target” or particular output, in this case, the final summary report. Depending on the computing environment, the essential processes will be carried out in an optimum manner.
Leveraging cutting-edge tool
For WGS-based TB molecular epidemiology, bioinformatics technologies are still under rapid development. However, there is disagreement about how they should be used in terms of both the choice of reference genomes and the requirement for recent transmission. TransFlow uses PANPASCO and TransCluster, two cutting-edge techniques, to address these issues. To prevent genetic distance computation bias, PANPASCO uses a pan-genome that represents the four major lineages (1-4) and a pairwise distance approach. In order to increase the detection rate of transmission clusters and the adaptability of samples, TransCluster is a revolutionary transmission cluster identification tool that incorporates sampling time, SNP distance, transmission rate, and molecular clock rate into its transmission probability model. Utilizing both technologies, TransFlow determines two reliable clusters (Cluster 1 and Cluster 4), which changes three clusters from the first CTB research.
Illustrating outcomes of the analysis
TransFlow generates charts or tables for every study so that users may quickly comprehend and use the findings. The most significant visualizations are all gathered into a single summary report file that emphasizes the key aspects of the study and details all the steps taken to produce the figure.
Employing snakemake as a framework
Snakemake, a scalable workflow engine that simplifies workflow management, serves as the foundation for TransFlow. The whole process is broken down into rules, each of which completes a single phase. The data flow is simple to follow since the output from one rule is the input for the rule that corresponds to the previous phase. TransFlow groups together in a single snakefile the rules that execute a significant portion of the workflow. The modules are all connected to the main Shell script through a shared configuration file. Users can use this script to run each module sequentially or to do an end-to-end examination. It is especially helpful when users wish to test out various factors, such as many clustering techniques.
Moreover, Snakemake deduces which rules may be executed concurrently and independently, minimizing CPU idle time and accelerating process completion. TransFlow allows users to choose between the tools used in the process since it is very modular and open source. For instance, to alter the sequencing reads alignment program from the default BWA to Bowtie2, the following procedures need to be carried out:
- Add Bowtie2 information to the Conda environment’s YAML file.
- Adjust a few elements of the Snakemake rule, which utilizes BWA for reading mappings, such as the output file format and shell instructions, to comply with Bowtie2’s specifications.
- Changes must be made to other rules that use these output files as input files.
Easily operable
Online instructions are available for installing, deploying, and utilizing TransFlow. It is important to note that TransFlow is made to work with the Bioconda channel and the Conda package management. This enables users to download and set up the several bioinformatics software programs that make up the system. With a single command, transFlow. With each option having a thorough description and suggested default value, the accompanying configuration file allows for fine-tuning of all applications and algorithms contained in TransFlow. Setting up a metadata file for TransFlow necessitates a basic understanding of how the terminal works and how to modify a TSV file using software like Excel.
Conclusions
TransFlow is a novel WGS-based TB transmission analysis pipeline that is quick, effective, adaptable, and simple to use, making it a valuable and cutting-edge tool for researchers. The entire operation begins with MTBC filtering and raw read quality control. Optional trimming, pangenome reference alignment, variant calling, pairwise SNP distance computation, transmission clustering, reconstruction of the transmission network, and risk factor inference are some of the processes it goes through.
Article Source: Reference Paper
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.
Riya Vishwakarma is a consulting content writing intern at CBIRT. Currently, she's pursuing a Master's in Biotechnology from Govt. VYT PG Autonomous College, Chhattisgarh. With a steep inclination towards research, she is techno-savvy with a sound interest in content writing and digital handling. She has dedicated three years as a writer and gained experience in literary writing as well as counting many such years ahead.





{kind=link}