
Scientists from Seoul National University, South Korea, and the Max Planck Institute for Multidisciplinary Sciences, Germany, have developed a deep learning-based method called Foldseek, which is a highly accelerated and accurate method for searching protein structures. Technological advances in structure prediction methods have yielded millions of protein structures, thus creating a bottleneck for searching operations in these databases. Foldseek describes tertiary amino acid interactions within proteins as sequences using a structural alphabet. This is how Foldseek aligns the structure of a query protein against a database. Foldseek has been shown to supersede Dali, TM-align, and CE by four to five orders of magnitude in terms of computation times.
Why do we need Foldseek?
The near-experiment quality protein structures that are being predicted using deep learning techniques such as AlphaFold2 have vastly advanced structural biology and bioinformatics. However, the pace at which structures are being reported is in great contrast to the computational time required to search through these databases. AlphaFold2 has already predicted 214 million structures found in the EBI repository. The ESM Atlas has over 617 million metagenomic structures predicted by ESMFold. The scale of such databases poses serious challenges to method development for navigating these databases and for performing analysis.
Sequence similarity search is the most widely used methodology for protein annotation and analysis, such as in MMseqs2, HH-suite3, and DIAMOND. Such annotation and analysis are based on the approach of finding homologous sequences to the query protein sequence from which properties such as cellular and molecular structures and functions can be inferred. However, it is often difficult to annotate proteins as detecting distant evolutionary relations from sequences alone is cumbersome.
The detection of similarity between protein structures using three-dimensional (3D) superposition offers greater sensitivity for identifying homologous proteins. High-quality structures are available for a vast number of proteins, and this information is easily incorporated for improving homology inference. However, structural aligners suffer from a lack of improved speed and sensitivity, and current aligner tools are not fast enough to cope with the scale of structural databases. Thus, a fast and accurate method for searching protein structures is needed.
How slow are the current structural tools?
The popular TM-align tool takes about a month’s time on one CPU to go through a database with 100 million protein structures. An all-versus-all comparison would require 10 millennia on a 1000-core cluster. In comparison, sequence searching is faster by an order of magnitude of four to five times. An all-versus-all comparison of 100 million sequences using MMseqs2 would take only about a week on the same cluster.
These structural alignment tools are slower for two main reasons. First, they do not incorporate prefilters, as is the case in sequence search tools, for gaining computational speed. Second, the structural similarity scores are non-local, meaning a change in alignment in one part affects the similarity in all other parts. Structural aligners such as TM-align, Dali, and CE use iterative or stochastic optimization to solve the alignment optimization problem.
How can the speed be improved?
The ground-breaking idea for increasing computation speed, as implemented in SA-Search, is that of describing the amino acid backbone of proteins as sequences over a structural alphabet and comparing structures using sequence alignments. Several approaches have been developed using this and by discretizing the local amino acid backbone, such as in CLePAPS.
The authors, however, used a different approach for reducing the computation speed for searching for protein structures. They developed a type of alphabet that does not describe the backbone but rather describes tertiary interactions within the protein.
Foldseek architecture: a brief overview
The structural alphabet devised describing tertiary interactions has 20 states. These 20 states of the 3D interaction (3Di) alphabet describe the geometric conformation for each residue i with its spatially closest residue j.
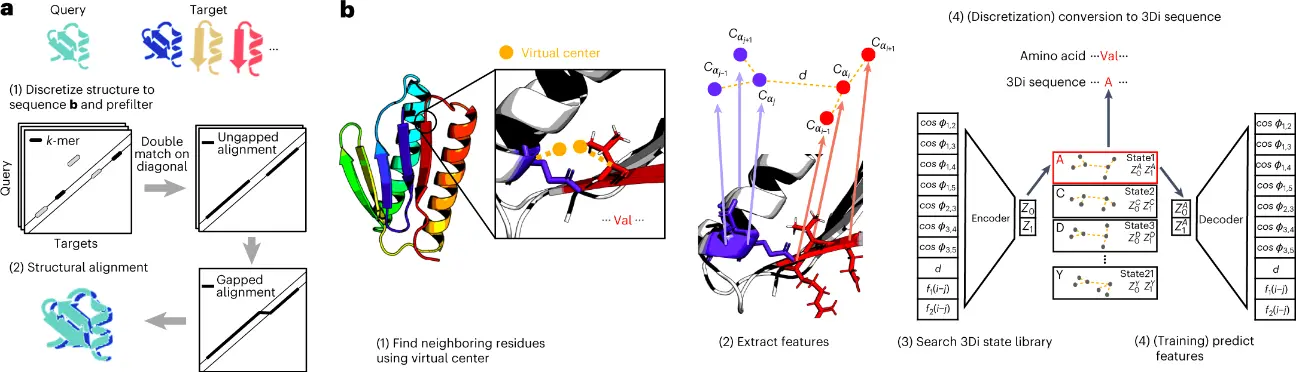
The Foldseek workflow is as follows:
- The method searches a set of query structures through a set of target structures by discretizing query and target structures into 3Di sequences.
- For the detection of candidate structures, the fast and sensitive k-mer and ungapped alignment prefilter of MMseqs2 is applied to the 3Di sequences.
- For learning the 3Di alphabet, ten features comprising Euclidean distance and seven angles and two sequence distance features describing the interaction geometry of residues i and j were used to define the 20 states of the 3Di states by training a VQ-VAE modified to learn maximally evolutionarily conserved states.
- For structure searches, the encoder predicts the best-matching 3Di state for each residue.
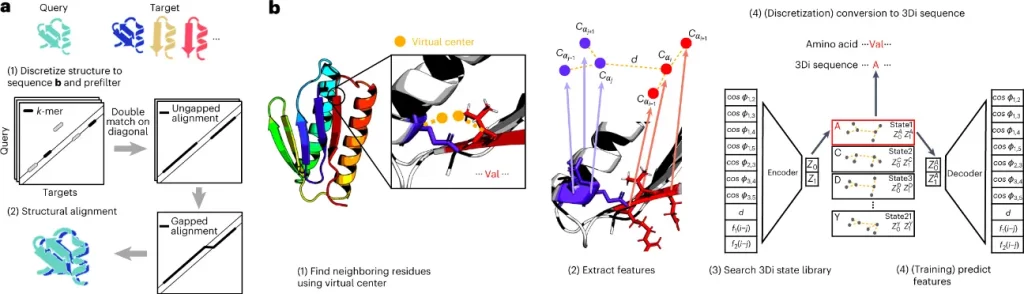
Image Source: https://doi.org/10.1038/s41587-023-01773-0
Conclusion
The authors developed a fast and highly accurate tool for protein structure search, Foldseek. They measured the speed and sensitivity of Foldseek by comparing it with other structural alignment tools and found it to perform with high speed and sensitivity. This is indeed a great tool in the toolbox of a structural biologist for keeping up with the pace of protein structure prediction. Thus, one can expect a fairly faster protein structure search protocol, which will remarkably aid the scientific as well as the drug development and therapeutic communities.
Article Source: Reference Paper | Foldseek: Webserver
Learn More:




Banhita is a consulting scientific writing intern at CBIRT. She's a mathematician turned bioinformatician. She has gained valuable experience in this field of bioinformatics while working at esteemed institutions like KTH, Sweden, and NCBS, Bangalore. Banhita holds a Master's degree in Mathematics from the prestigious IIT Madras, as well as the University of Western Ontario in Canada. She's is deeply passionate about scientific writing, making her an invaluable asset to any research team.





{kind=link}