
Scientists from the University of Michigan developed an open-source program, DeepFold, to quickly construct accurate protein structure models from deep learning-based potentials.
In spite of recent progress in protein structure prediction, it is still necessary to improve the accuracy of models for proteins lacking sequence and/or structure homologs. The DeepFold program uses multi-task deep residual neural networks and knowledge-based energy functions to guide its gradient-descent folding simulations based on spatial restraints.
A large-scale benchmark test demonstrated that DeepFold could create full-length models with significantly greater accuracy than classical folding approaches and other leading deep learning approaches. It is noteworthy that DeepFold achieved a TM-score that was 44.9% higher than DMPfold on the most difficult targets with very few homologous sequences. A DeepFold simulation was 262 times faster than a traditional fragment assembly simulation. Using accurately predicted deep learning potentials, ab initio protein structure prediction can be improved both in accuracy and speed.
Using a protein’s primary sequence, structure prediction aims to determine every atom’s spatial location. It is possible to divide protein structure prediction methods into template-based modeling (TBM) and template-free modeling (FM), the latter of which is also called ab initio modeling, based on the availability of reliable structural templates in the PDB. While TBM has been the most reliable method for modeling protein structures for many years, its accuracy depends largely on close homologous templates and matching query-template alignments. For proteins lacking homologous templates in the PDB, ab initio methods are designed to improve folding performance by using advanced energy functions and sampling techniques. Although physics-based FM methods have improved significantly for targets with readily identifiable homologous templates, their performance for non-homologous targets has remained significantly inferior as a result of inaccuracies in force field design and limitations of conformational search engines.
As deep learning techniques have been applied to predict spatial restraints from sequences and/or multiple sequence alignments (MSAs), ab initio structure prediction has dramatically improved. Specifically, in CASP11 and CASP12, predictors primarily used direct coupling analysis from MSAs and shallow neural networks to forecast contact maps. In order to accurately predict contacts based on information from correlated mutation patterns, the prediction accuracy largely relied on the identification of abundant sequence homologs. For example, Zhang-Server and QUARK, the top-ranked server groups in CASP13, used contact maps derived from deep convolutional residual networks (ResNets) to guide I-TASSER and QUARK folding simulations, respectively, resulting in improved contact prediction and folding accuracy for physics-based modeling and knowledge-based modeling methods.
In particular, homologous templates and high-quality MSAs were lacking for targets without homologous templates. Improved protein sequence matching can also yield more structure contacts, which can also be used to improve protein sequence matching. A number of deep learning constraints were integrated into the folding simulations in CASP14, the most recent CASP experiment. These include distance maps, which are conceptually similar to contact maps, but also contain information about inter-residue distances, dihedral angles between residues, and hydrogen bonds. Due to the introduction of more precise spatial information to guide the folding simulations, the results demonstrated significant improvements over the contact-based structure assembly approaches.
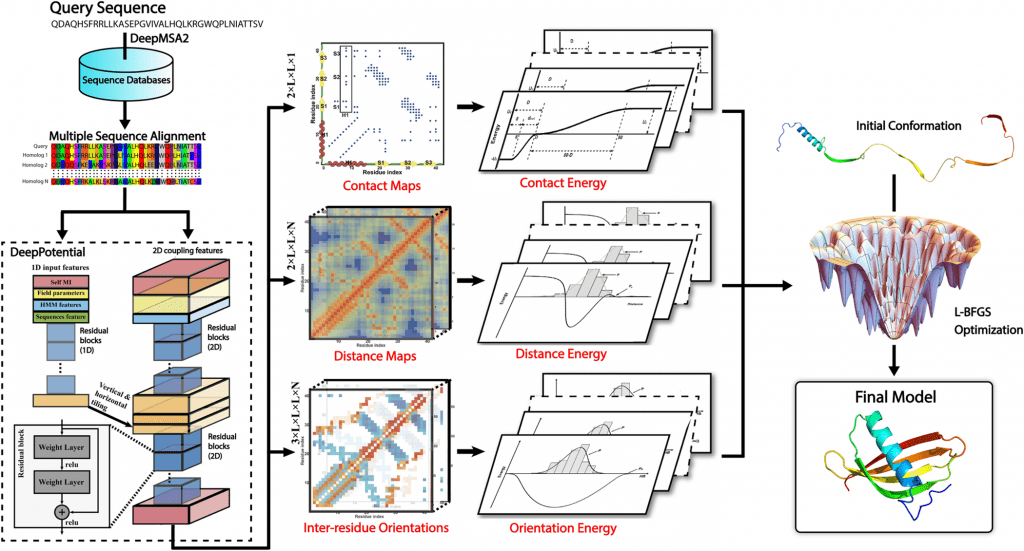
Image Source: https://doi.org/10.1371/journal.pcbi.1010539
Although modeling accuracy has improved, approaches based on fragment/template assembly folding techniques, including I-TASSER, Rosetta, and QUARK, often require long simulation times, especially for longer proteins, which makes them unsuitable for large-scale modeling. Protein folding involves an enormous structure space and a complex energy landscape, requiring extensive conformational sampling for ab initio modeling.
Advanced deep learning techniques now provide abundant high-quality restraints, even when paired with sparse spatial constraints from threading alignments and low-resolution experiments. A large amount of energy can be smoothed out by these abundant and accurate restraints. A large number of folding simulations may no longer be necessary in this regard, which partly explains the remarkable success of other CASP teams like AlphaFold in CASP13 and TrRosetta in CASP14, which used conformational searching procedures based on local gradient descent.
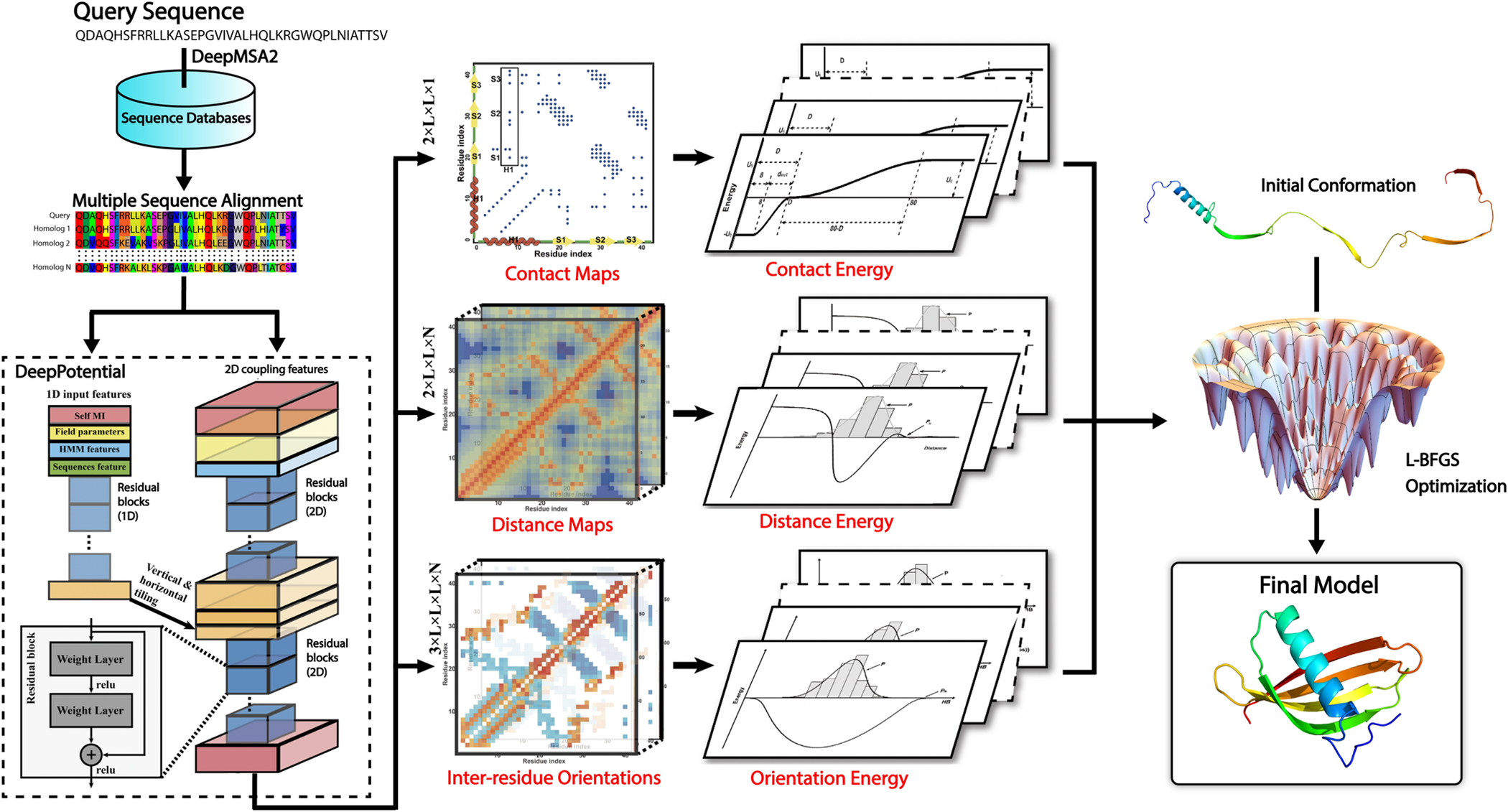
By combining a general knowledge-based statistical force field with a deep learning-based potential produced by the DeepPotential program, an open-source protein folding pipeline, DeepFold, increases the accuracy and speed of ab initio protein structure predictions has been developed. Compared to traditional folding simulation methods, the pipeline demonstrated superior performance on large-scale datasets compared to other leading structure prediction approaches. L-BFGS structure optimization pipeline and deep learning models are all included in the program.
Using DeepMSA2, DeepFold creates an MSA by searching multiple whole genome and metagenomic databases. A deep ResNet architecture within DeepPotential uses the coevolutionary couplings derived from the MSA as input features to predict spatial restraints, including distance/contact maps and inter-residue torsion angle orientations. As a result of converting the restraints into a deep-learning-based potential, the L-BFGS folding simulations for generating full-length models are guided by this potential, along with a general knowledge-based physical potential.
To improve MSA quality and reduce the time required to search the various sequence databases, more efficient and precise MSA construction strategies should also be developed. As the size of sequence databases, primarily metagenomics databases, grows, increasing search efficiency is especially crucial. In addition to allowing more sequences to be collected, it also increases the time and computational resources needed for searching the sequence databases, increasing the possibility of false negative sequence samples. By focusing on search sequences related to the target protein’s biome, a targeted MSA generation protocol can significantly increase the speed and quality of MSA generation, thus improving the accuracy of the final 3D structure model.
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.
Srishti Sharma is a consulting Scientific Content Writing Intern at CBIRT. She's currently pursuing M. Tech in Biotechnology from Jaypee Institute of Information Technology. Aspiring researcher, passionate and curious about exploring new scientific methods and scientific writing.





{kind=link}