
The algorithm is the first full-fledged machine learning-based short read alignment algorithm to speed up DNA sequencing, that efficiently uses learned indices to solve the exact match search issue for efficient seeding.
The human genome is made up of a whole set of DNA that is around 6.4 billion letters long. Reading the entire genome sequence at once is difficult due to its vastness. As a result, scientists employ DNA sequencers to generate hundreds of millions of DNA sequence fragments, or short reads, with lengths of up to 300 letters. The DNA sequencer then uses a huge jigsaw puzzle to piece together all of the short readings to rebuild the whole genome sequence. Even with the most powerful computers, this task can take many hours to accomplish.
A KAIST research team developed the first short-read alignment software that employs a recent advancement in machine learning termed a learned index to reach up to 3.45x quicker rates.
The study team published the findings in the journal Bioinformatics in March 2022. The open-source software ‘BWA-MEME’ is available at https://github.com/kaist-ina/BWA-MEME.
NGS (next-generation sequencing) is a cutting-edge DNA sequencing technique. Projects to achieve population-scale genomic sequencing are now underway. Modern NGS hardware may generate billions of short reads in a single run. Following that, the short reads must be aligned to the reference DNA sequence. The necessity for an effective short read alignment tool has become even more crucial as large-scale DNA sequencing operations run hundreds of next-generation sequences. Accelerating the alignment of DNA sequences would be the first step toward population-scale sequencing.
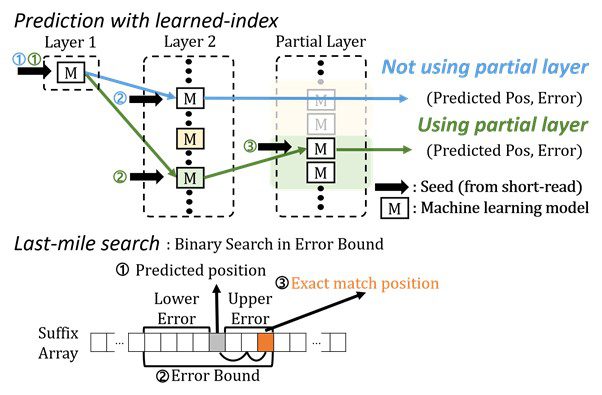
The short-read alignment software tool BWA-MEM2 is now being utilized to sequence the DNA. It does, however, have certain restrictions. Seeding and extending are the two processes of cutting-edge alignment. Searches for exact matches of short reads in the reference DNA sequence are performed during the seeding phase. The brief reads from the seeding phase are stretched during the extending phase. The seeding phase of the existing process has bottlenecks. The search for specific matches slows down the procedure.
The most challenging part of the seeding process is finding accurate matches of short-read substrings in the reference DNA sequence. The existing methods have performance limitations due to frequent memory accesses.
The researchers set out to discover a means to speed up the alignment of DNA sequences. They used machine learning approaches to make an algorithmic upgrade to speed up the procedure. BWA-MEME (BWA-MEM emulated) approach uses learned indices to tackle the exact match search problem. For an exact match search, the original software compared one character at a time. By lowering the number of instructions by 4.60x and memory accesses by 8.77x, the team’s novel technique achieves up to 3.45x quicker seeding throughput than BWA-MEM2.
It has been demonstrated in the study that using machine learning technology, entire genome big data analysis can be accomplished faster and cheaper than traditional approaches.
Prof. Dongsu Han, KAIST’s School of Electrical Engineering
The researchers’ ultimate goal was to create efficient software for processing massive data in genomics that experts from academia and industry could use on a regular basis.
We see so many chances for building better software for genetic data processing with recent breakthroughs in artificial intelligence and machine learning. The potential for speeding up existing analysis and enabling new forms of analysis exist, and our goal is to develop such software.
Prof. Dongsu Han
Whole-genome sequencing has long been used to find genomic alterations and the origins of diseases, leading to the finding of new drugs and therapies. There could be a wide range of applications. The analysis of whole-genome sequences is used not only in research but also in clinical approaches. The science and technology for evaluating genetic data are rapidly advancing, making it more accessible to researchers and patients. This can help us better understand diseases and offer better treatments for patients with diverse diseases.
Story Source: Youngmok Jung, Dongsu Han, BWA-MEME: BWA-MEM emulated with a machine learning approach, Bioinformatics, Volume 38, Issue 9, 1 May 2022, Pages 2404–2413, https://doi.org/10.1093/bioinformatics/btac137
https://news.kaist.ac.kr/newsen/html/news/?mode=V&mng_no=20490
Dr. Tamanna Anwar is a Scientist and Co-founder of the Centre of Bioinformatics Research and Technology (CBIRT). She is a passionate bioinformatics scientist and a visionary entrepreneur. Dr. Tamanna has worked as a Young Scientist at Jawaharlal Nehru University, New Delhi. She has also worked as a Postdoctoral Fellow at the University of Saskatchewan, Canada. She has several scientific research publications in high-impact research journals. Her latest endeavor is the development of a platform that acts as a one-stop solution for all bioinformatics related information as well as developing a bioinformatics news portal to report cutting-edge bioinformatics breakthroughs.











{kind=link}