
AlphaFold, a machine learning system developed by Google DeepMind, can predict the three-dimensional structures of proteins with unprecedented accuracy, leading to a better understanding of how these structures relate to function. The researchers from USDA-ARS describe a methodology “Functional Annotations using Sequence and Structure Orthology (FASSO),” which takes advantage of AlphaFold along with sequence and structure orthology to assign functional annotations to proteins accurately.
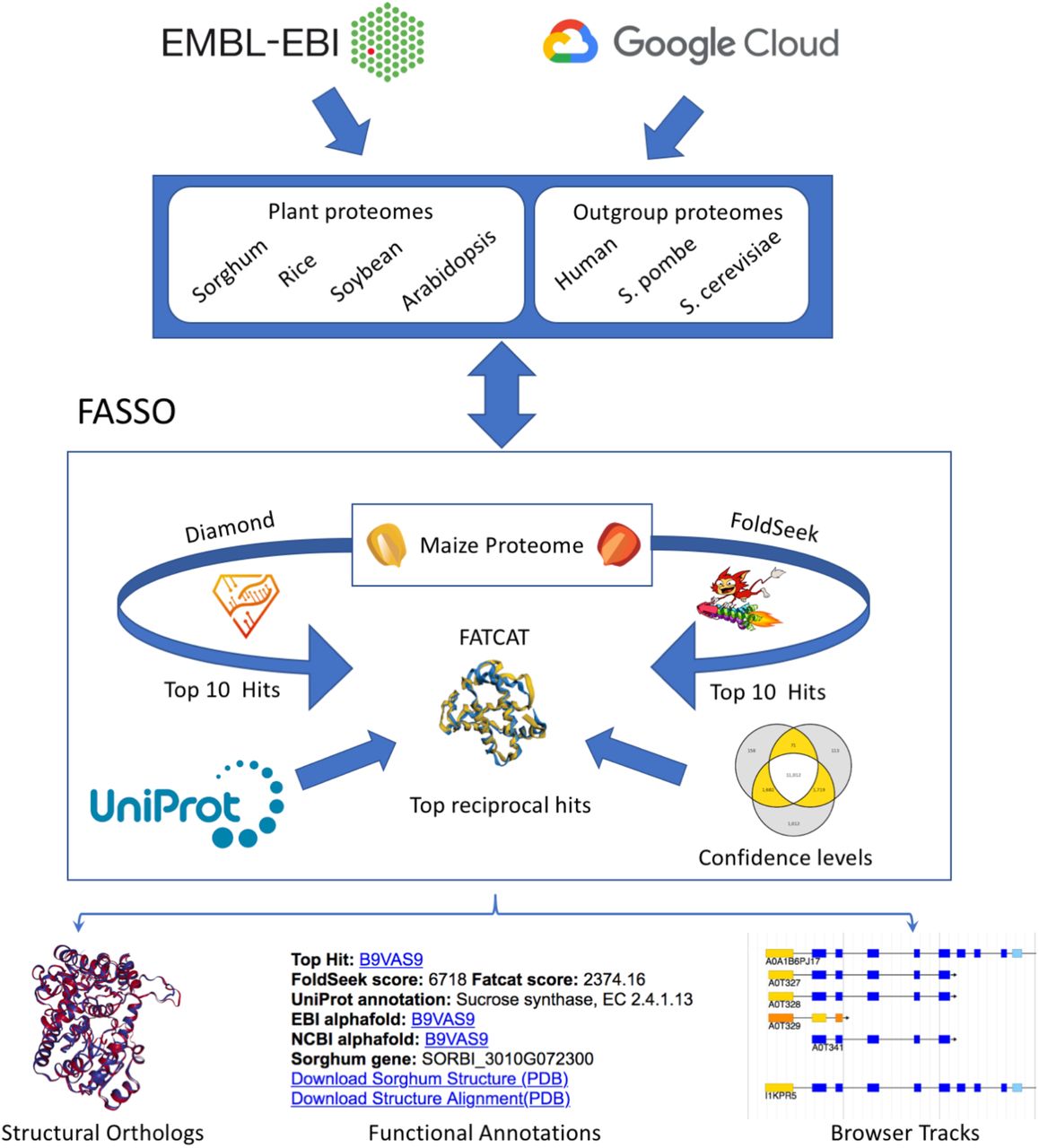
FASSO uses both protein sequence and structure information to enhance the precision of proteome-scale functional annotation operating in a wide spectrum of taxa by increasing the number of high-quality predictions and removing potentially incorrect annotations.
Proteome comparison and annotation require the ability to compare and annotate proteomes rapidly and precisely, which is essential for organizing experiments, making critical research choices, and developing novel ideas. Annotation of orthologs between five plant species (maize, sorghum, rice, soybean, Arabidopsis) and three distance outgroups (human, budding yeast, and fission yeast) has been carried out using FASSO.
Sequence and Structure-based Approaches for Functional Annotations
Annotation of newly sequenced organisms, functional genomics, gene organization in species, evolutionary studies of biological systems, and phylogenomic analyses demonstrate Ortholog detection’s significance in comparative genomics and bioinformatics. This steers the use of model organisms to study conserved processes such as cell cycle progression, DNA replication, and transcription that are more difficult to assay in complex systems based on sequence similarity with other genomes with annotated functions. Sequence similarity is not necessarily a good criterion for predicting protein function, given that distinct amino acid sequences can produce identical structures and vice versa.
Reciprocal Best Hits (RBH), which assumes orthologs if two genes from separate genomes each identify the other as the best hit in the other genome, is one of the shortcuts needed for the analysis of the growing numbers of genome sequences.
Image source – doi: https://doi.org/10.1101/2022.11.10.516002
The most commonly used program for finding RBH sequence matches is blastp, blat, lastal, Diamond, and MMseqs2. Diamond offers a good compromise in speed, sensitivity, and quality.
Less than ten percent of protein pairings have structural similarity, which is commonly referred to as the “twilight zone” in couples with a sequence identity of 20 to 35 percent. As evidenced by the experimentally negligibly low number of solved protein structures (44,000 entries in the Protein Database (PDB)) compared to the number of known protein sequences (over 200 million protein sequences in the UniProt database), it is simpler and less expensive to determine protein sequences than structures.
However, recent developments allow for the precise prediction of protein structures for any amino acid sequence using methods such as RoseTTAFold, OmegaFold, and ESMFold.
Potential new orthologs between humans, worms, fruit flies, and yeast were discovered using the reciprocal best structure hit technique (RBSH) and FoldSeek.
This approach has been extended by aggregating predictions from a sequence-based approach (Diamond) with two structure-based approaches (FoldSeek and FATCAT) to identify structural orthologs, assign functional annotations, and flag conflicting predictions. Functional Annotations using Sequence and Structure Orthology (FASSO) identify structural orthologs and transfer functional annotations for eight diverse proteomes that include five plants (maize, sorghum, rice, soybean, and Arabidopsis) and three well-annotated outgroups (human, S. cerevisiae, and S. pombe).
At least two proteomes with PDB-formatted structures are necessary for the FASSO process. A data preparation script is offered to extract the data, construct the directory structure, format the data, and generate databases and indexes for further steps.
Each protein in the target proteome is aligned with the query proteome via Reciprocal Best Hit (RBH) approach (Diamond).
Reciprocal Best Structure Hit (RBSH) is a method used in FoldSeek to find protein couples from various proteomes that have the best structural alignments. Proteins are referred to as structural orthologs if their top structural alignments are with one another. Between proteomes, FoldSeek structurally aligns the proteins. FATCAT is used in the second round of RBSH. By enabling twists around pivot points in the structures, FATCAT differs from FoldSeek. Aligned fragment pairs (AFP) between the two proteins are chained together by FATCAT through the use of dynamic programming.
Predicting Potential Orthologs
The reciprocal top performances from Diamond, FoldSeek, and FATCAT are combined in FASSO’s last step. Based on the consensus among the three approaches, each ortholog is given a confidence label of platinum, gold, or silver.
- If all methods predict the same ortholog pair – Platinum;
- If two of the three methods agree – Gold;
- If only a single approach makes the prediction – Silver.
For downstream analysis, the final ortholog predictions for the maize genome were aligned back to the B73 maize reference genome. The query database was built using the genomic coordinates.
Outcomes of FASSO
The final FASSO output provides the following:
- A set of structural orthologs with confidence assignments
- Functional annotations for each protein
- A working set of flagged orthologs
AlphaFold database of three well-annotated proteomes as outgroups: Homo sapiens (human), Saccharomyces cerevisiae (S. cerevisiae, budding yeast), and Schizosaccharomyces pombe (S. pombe, fission yeast).
Maize genome has been used as a reference since it is –
(1) An important agronomic crop and scientific model organism ;
(2) A model plant species with a wealth of genetic, genomic, and phenomic datasets and resources;
(3) Well studied with a few hundred well-annotated genes but has limited experimentally determined functional annotations at the genome-scale; and
(4) Has annotated genomes at multiple levels of taxonomic distances.
Three criteria are used to rate the accuracy of predicted protein structures on a proteome scale:
- Residue quality ratio.
- Average residue score across all the proteins in each proteome.
- Distribution of the average per-residue score per protein.
The efficiency analogy of Diamond, FoldSeek, and FATCAT can be seen on an HPC, FoldSeek concluded its run in less than a day, whereas Diamond finished the all-vs-all run in a matter of minutes. Running an all-vs-all strategy is not rational since FATCAT can take up to a day to compare a single protein to a proteome.
Projecting Orthologs via FASSO and Reciprocal Best Hit Results
Each method’s results are combined by FASSO, which also provides confidence labels depending on the amount of agreement and eliminates predictions that contradict each other. Another issue is when there is a significant sequence or structural similarity between localized portions of distinct proteins, yet the overall similarity may be extremely different. By offering bi-directional support for a prediction, the reciprocal best hit method partially resolves this issue, while FASSO, which combines sequence-based and structure-based predictions, offers even better support.
Annotating the Maize Proteome
The 243,292 proteins in the eight proteomes received 2.1 million functional annotations from Diamond, FoldSeek, and FATCAT. To create a final collection of 271,278 functional annotations, FASSO combined these findings and eliminated predictions that conflicted with each other.
Uncharacterized protein, often known as hypothetical protein, is a general term used to annotate proteins having uncertain or missing activities. These proteins frequently exhibit open reading frames, gene expression, and/or conserved homology, which are all indications that they have a functional purpose. They still require supporting data to be given a specific function. Researchers looking for important hints while exploring causative genes may need help with these annotations. In a well-annotated proteome, the proportion of uncharacterized proteins can range from a small number to more than 50%.
Using FASSO, new functional annotations to 1,021 of the 9,103 uncharacterized proteins in maize are allotted.
Summing-Up
Sequence similarity has long been utilized in bioinformatics to establish orthologous connections and deduce biological functions. Sequence similarity remained an unreliable indicator of functional similarity in the absence of proteome-scale protein structural knowledge.
FASSO is already touch-stoned by employing more than 170 billion protein alignments to predict structural orthologs and assign functional annotations for 64 pairings of proteomes from eight different species.
Article Sources: Reference Paper
Learn More:
Top Bioinformatics Books ↗
Learn more to get deeper insights into the field of bioinformatics.
Top Free Online Bioinformatics Courses ↗
Freely available courses to learn each and every aspect of bioinformatics.
Latest Bioinformatics Breakthroughs ↗
Stay updated with the latest discoveries in the field of bioinformatics.
Riya Vishwakarma is a consulting content writing intern at CBIRT. Currently, she's pursuing a Master's in Biotechnology from Govt. VYT PG Autonomous College, Chhattisgarh. With a steep inclination towards research, she is techno-savvy with a sound interest in content writing and digital handling. She has dedicated three years as a writer and gained experience in literary writing as well as counting many such years ahead.





{kind=link}