
Tuberculosis has been known to be a significant threat for many years now. Despite the development of a number of preventative strategies and medications, it remains widespread, causing millions of deaths globally every year. Part of the difficulty of adequately responding to and treating tuberculosis is due to the emergence of strains that are resistant to drugs. The ability to detect genetic markers of antimicrobial resistance within Mycobacterium tuberculosis, as well as other pathogens, would be of significant use in trying to combat this deadly disease—and a group of scientists from Saarland University have developed a new bioinformatics method to do just that.
New advances in biology over the course of the last century have made it possible for humanity to combat various deadly diseases. Indeed, some of these battles were won so decisively that some diseases are now considered to be eradicated in many places. However, the injudicious use of antibiotics and other drugs has led to the development of antimicrobial resistance in various pathogens—leading to the emergence and spread of so-called “superbugs.” This has been a cause for significant concern among medical and public health organizations for decades now and has been the subject of much research.
Drug resistance was detected in 4.5% of more than 10 million tuberculosis cases reported by the World Health Organization in 2019. Prior research into Mycobacterium tuberculosis (Mtb) – the pathogen responsible for tuberculosis – has revealed that genetic mutations like insertions, deletions, and SNPs (single nucleotide polymorphisms) are significant in inducing antimicrobial resistance to many tuberculosis drugs.
Computational Methods to Identify Resistance-Associated Mutations
Bioinformatics has been instrumental in transforming the field of genetics research by making it easier and faster to analyze large amounts of data. Whole genome sequencing (WGS) has been increasingly utilized to identify mutations that cause resistance. Methods like phylogenetic convergence testing and identifying genes found only in resistant variants have been successfully used to discover resistance-causing mutations. Genome wide association studies (GWAS) are also commonly used for this purpose. Information about bacterial mutants can mainly be garnered from genomic sequences. Bacteria tend to have long genome segments, strong linkage disequilibrium, not to mention long accessory genomes and loci possessing different allelic variants. M. tuberculosis stands apart from other bacterial species due to its low recombination rates and comparatively tiny accessory genome.
As M. tuberculosis’ loci are nearly all in a state of linkage disequilibrium, it is clear that population structure is very significant, which should be considered when conducting genome-wide association studies. The use of linear mixed models accounts for the fact that any variants will also have multiple passenger mutations associated with them due to different strains being closely related. Linear mixed models model the impact of a particular locus and contextualize it with that of other loci to systematically decrease the effect of a given locus that correlates to the background. This method has been used successfully for different bacterial species, including S. aureus, E. coli, K. pneumoniae, etc.
Using genome-wide association studies in tandem with phylogenetic convergence testing allowed researchers to identify how genes associated with drug resistance interacted with each other. This approach considers the theory of driver mutations that confer resistance originate independently in different branches, while passenger mutations only occur in individual branches.
Other factors, such as polygenicity, multi-allelic SNPs, recombination rate, and more, should also be accounted for in GWAS. Multiple tools were developed for this purpose, like CCTSWEEP, treeWAS, Scoary, and SEER, among others. However, many of these don’t consider the obstacles that GWAS may encounter. Other methods, like the TB profiler, call out certain variants and compare them to those in databases. This means resistant variants can only be detected if they possess known resistance-conferring mutations. Those strains whose antimicrobial resistance is caused by as-yet-unknown mutations will pass by undetected. Machine learning approaches have been used to rectify this and provide a promising new avenue for further research. They have been employed to detect resistance against certain drugs in M. tuberculosis. A framework, TB-ML, was recently developed and provides the capability to implement various common machine learning methods, like direct association, convolutional neural networks, and random forests. However, these models don’t consider bacterium-specific features, like linkage disequilibrium or population structure. Hence, the ranking of variants must be done before the application of ML models so that insignificant features can be filtered out.
The Introduction of a New Method – Phylogeny-related Parallelism Score
A new method has been developed to rank genetic variants and train machine-learning models to predict antibiotic resistance in M. tuberculosis. A dataset of more than 4000 genome sequences was used. These sequences were then mapped to the reference genome (H37Rv). Variants that were only present in <0.2% of the strains were removed, as were INDELS, leaving more than 24,400 SNPs that were used for further study. The PRPS (phylogeny-related parallelism score) was calculated, so mutations strongly associated with population structure were excluded. The approach led to the successful identification of multiple mutations known to confer resistance, including for ofloxacin and aminoglycoside resistance.
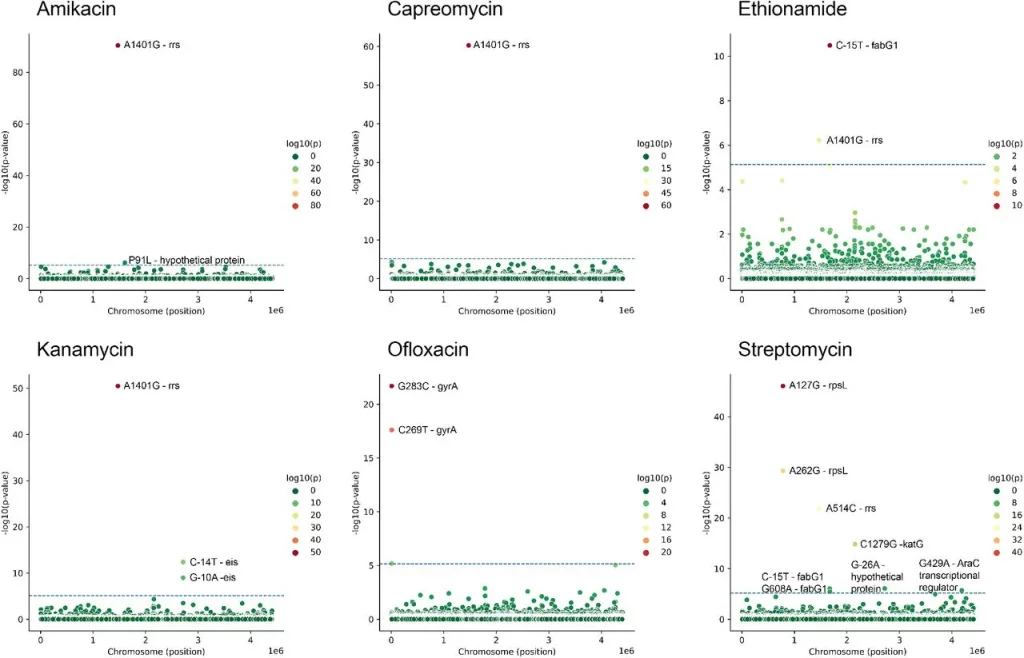
Image Source: https://doi.org/10.1101/2023.09.06.556328
Associations between SNP and antibiotic resistance were found using GWAS (see Methods). Horizontal line shows selected threshold for significance.
SNPs were observed upstream of many resistance-associated genes, indicating that regulation of their expression also played an important role in conferring resistance. Furthermore, new associations were observed between genes that conferred resistance to certain antibiotics and resistance to other drugs as well. For example, EmbA is associated with ethambutol resistance but can also potentially predict ethionamide resistance, indicating multi-drug resistance.
Additionally, new mutations were described as having an association with resistance. When analyzed with AlphaFold, it was found that some of these mutations, such as Ile145Met in PabC and Lys179Gln in FtsH, are located on a protein-protein interaction interface, indicating there is a significant change in certain interactions that may cause resistance to be conferred. The latter gene, FtsH, has also been found to be associated with antimicrobial resistance in other bacteria, like P. aeruginosa and S. aureus.
Conclusion
The use of PRPS to rank variants was demonstrated to have a positive impact on the models’ performance and was shown to be able to minimize the number of false positives. This helps rectify some of the pitfalls associated with the commonly used WGS method, namely the high incidence of false positives due to strong population structures and relatively low recombination rates.
While this study restricted itself to SNPs, other mechanisms, like plasmid gene transfer, chromosomal rearrangements, and gene copy numbers, can also be incorporated into machine learning models. Additionally, phenotypic screens of the kind that were used to train the machine learning models can be quite expensive; as such, the development of suitable algorithms that can work with limited data is also necessary.
Despite these drawbacks, the development of such a model is demonstrably useful and shows the utility of machine learning and other computational tools in biological research. Garnering similar results through wet lab experimentation can be time-consuming and expensive, hence, the use of such bioinformatics tools to optimize and quicken the process can be beneficial.
Article Source: Reference Paper
Learn More:




Sonal Keni is a consulting scientific writing intern at CBIRT. She is pursuing a BTech in Biotechnology from the Manipal Institute of Technology. Her academic journey has been driven by a profound fascination for the intricate world of biology, and she is particularly drawn to computational biology and oncology. She also enjoys reading and painting in her free time.











{kind=link}
Thanks for your support.
Good work. i would like to learn more about the new Bioinformatics tools.