
The molecular surface of a protein is represented in the form of geometric and chemical attributes that create the unique fingerprint for identifying protein interaction. Geometric deep-learning-based algorithms can be used to analyze these fingerprints, which can help researchers design new computational tools to study Protein interactions.
Inferring protein-protein interactions (PPIs) solely from their structural information remains a major issue in the field of proteomics. The unavailability of tools and techniques to understand the mechanism of protein interaction has been a great hindrance in designing antibodies and regents with an affinity for selected target proteins.
Protein-Protein Interactions
One of the problems faced by researchers studying protein-protein interactions is generating a protein sequence that can bind to the target protein and form a complex structure. The synthesized protein sequence is known as a protein binder. The availability of several tools like AlphaFold has helped researchers design and predict protein-protein interactions to some extent. But protein binder design is a relatively less researched field.
Scientists have tried to overcome this problem by developing a neural network-based tool that considers proteins as surfaces and learns from the geometric and chemical attributes present on the protein surface. It can determine the probability of an interaction between two protein molecules. The recognition of a molecular surface is based on its geometric and chemical constraints, which are interdependent but reciprocal in nature. Geometric constraints result from van der Waals interactions, and chemical constraints arise due to hydrophobic and electrostatics interactions within the protein complex structure.
It has been observed that most proteins contain a core interface that is buried inside the protein complex structure and makes up the surface regions that are not accessible to solvent. Interface rims are patches of proteins exposed to solvent. Some residues were observed to be more susceptible to mutation and contribute towards protein-protein interactions. They were referred to as “hotspots.” Based on these findings, the researchers have come up with an approach based on the importance of these buried core interfaces. They have utilized surface fingerprints from the surfaces of interacting proteins to predict probable sites for protein interactions. This approach is fast and accurate in predicting sites for protein interactions and designing protein binders. The researchers using this technique have been able to design protein binders for four target proteins: PD-1, PD-L1, CTLA-4, and the spike protein of SARS-CoV-2.
Steps followed in designing new protein binders:
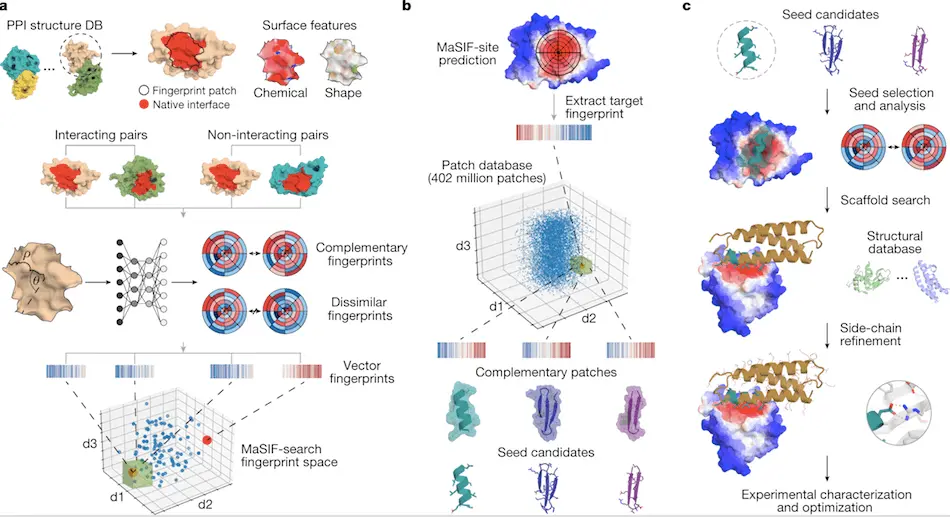
1. The primary step in this process is surface fingerprint generation using the MaSIF tool. This includes the identification of buried sites on the surface of interacting proteins using the MaSIF-site tool.
2. The second step includes the identification of new binding seeds by surface fingerprint-based search using the MaSIF-seed tool.
3. The last step includes transferring binding seeds into the protein scaffold and remodeling the protein binding interface using Rosetta to generate binder proteins. Best binder proteins are selected and tested experimentally.
Prediction of Protein Interaction Sites using MaSIF
The researchers have already released a tool based on a geometric deep-learning approach for identifying surface fingerprints called MaSIF (Molecular Surface Interaction Fingerprinting). It is also trained to predict the probability of an area on the surface of a protein generating a protein-protein interaction. It generates output by assigning probability scores for each surface point, indicating the possibility of becoming a buried site within a protein-protein interaction. The MaSIF tool is used to predict and design protein-protein interactions based on the structural information of the target protein. The working principle of the MaSIF tool is as follows: 1) Predicting buried interfaces of the target protein with a higher probability of binding. 2) Similar structural motif search using Surface fingerprinting (concept of MaSIF seed/binding seed) and 3) Placement of the structural motif into protein scaffolds for increasing stability of the protein interaction.
MaSIF-seed
The MaSIF-seed—protocol is a set of steps followed to identify new binding seeds within protein-protein interactions. Binding seeds refer to similar structural motifs that can bind to the target sites. MaSIF-seed breaks down the molecular surfaces of proteins into intersecting circular patches. Geometric and chemical attributes are calculated for each point inside these patches. Surface fingerprints are then generated using a Neural Network, which groups the output based on protein interaction. Similar fingerprints are generated for interacting proteins, and dissimilar fingerprints for non-interacting proteins. The fingerprints thus generated are aligned to the target sites, and the result was fed to a second neural network. This generates Interface Post-Alignment (IPA) score for the surface fingerprints. The outputs generated by MaSIF-seed were termed binding seeds and acted as substrates for designing protein binders for target proteins.
The MaSIF-seed tool’s precision and speed in accurately differentiating probable protein binders from decoy ones based on protein surface attributes helped the researchers design new protein binders that can be used to target proteins involved in diseases. The scientists were able to design binders to target the receptor binding domain of the SARS-CoV-2 spike protein. They also successfully designed binders for the CTLA-4 target protein, owing to its importance in immunology. Researchers also designed a protein binder for the PD-1-PDL1 complex, an important interacting protein relevant to oncology.
MaSIF-search
The researchers also released a search tool based on a Siamese neural network to examine surface constraints between two binding proteins known as MaSIF-search. The complex architecture of the tool was trained to generate identical fingerprints for the target/binder patch and dissimilar fingerprints for the target/random patches.
MaSIF-site
MaSIF-site, another tool developed by the researchers, was used to predict areas with high affinity to form protein-protein interactions. It generates a score based on the probability of each point forming a buried surface in a protein interaction. The MaSIF-site tool was trained using databases of protein-protein interactions like PRISM, ZDock benchmark, SabDab, and PDBBind.
Conclusion
The recent COVID-19 pandemic compelled scientists and researchers to develop advanced technologies for the early detection, prediction, and transmission of the disease. One such field was the proteomics-based study of the SARS-CoV-2 spike protein, which was undergoing rapid mutation. The spike protein was used to identify the highly transmissible COVID-19 from the flu. The researchers designed a surface-based protein binder that was able to bind the receptor-binding domain of the SARS-CoV-2 spike protein. This helped in the early detection of COVID-19 from the biological sample collected from patients and was crucial in limiting disease transmission. Scientists also developed protein binders for PD-1, PD-L1, and CTLA-4 for their clinical significance. Experiments proved that site-specific protein binders could be designed using different structurally similar motifs using computational tools.
Article Source: Reference Paper | MaSIF-seed: Github Link
Learn More:




Sipra Das is a consulting scientific content writing intern at CBIRT who specializes in the field of Proteomics-related content writing. With a passion for scientific writing, she has accumulated 8 years of experience in this domain. She holds a Master's degree in Bioinformatics and has completed an internship at the esteemed NIMHANS in Bangalore. She brings a unique combination of scientific expertise and writing prowess to her work, delivering high-quality content that is both informative and engaging.





{kind=link}