
Molecular biology is being revolutionized by the application of artificial intelligence to simulate the structures of proteins and their complexes. Researchers utilized a combination of in-cell crosslinking mass spectroscopy and co-fractionation mass spectroscopy in order to find protein-protein interactions in Bacillus subtilis. With the help of AlphaFold-Multimer, the scientists predicted the structures of protein interactions, including those in the SubtiWiki database, and after controlling for false-positive results, 153 dimeric and 14 trimeric protein assemblies in total were proposed as new structural models. This method provides structural insight into protein interactions within intact cells and is relevant to genetically resistant species, such as harmful bacteria.
Exploring Protein-Protein Interactions
Protein-protein interactions (PPIs) are essential for the operation of living organisms, yet their structures are infamously hard to understand. Proteomic and genetic methods have been utilized to create lists of PPIs, however, they provide limited insight into their topology.
The recent development of AlphaFold-Multimer has enabled precise predictions of the structures of protein-protein complexes, making it feasible to establish structure-function correlations across whole interactomes and possibly address the problem of understudied proteins. But, predicting the interaction interfaces of all potential protein pair combinations is computationally unfeasible, especially when bigger protein complexes with numerous subunits are considered. Thus, scientists are interested in identifying the topology of protein interactions without depending on tedious experimental methods.
Limitations of the existing methods and Crosslinking MS
In order to find protein-protein interactions (PPIs) in biological systems, experimental methods such as AP-MS and CoFrac-MS have been utilized. Nevertheless, these methods give minimal topological or structural information, usually leaving even the directness or indirectness of an interaction unclear. These methods may miss weak connections lost during cell lysis and interactions dependent on the cell’s original environment.
In recent years, in vivo crosslinking of proteins followed by identification of the connected residue pairs by mass spectrometry (crosslinking MS) has developed as a technology that can identify PPIs in cells and offer topological information about these interactions. It gives insights into the complex structure of protein-protein interactions in their original environment by fixing interactions within cells as the initial phase of the analytical workflow and providing information on linked residue pairs. However, it is uncertain if this approach can capture connections easily lost during cell lysis.
Discovering PPIs and Predicting Structures
Scientists present a novel approach for discovering protein-protein interactions (PPIs) in B. subtilis, a Gram-positive bacteria. They employ two experimental approaches, crosslinking mass spectrometry (MS) and co-fractionation MS, to capture PPIs within cells and offer topological information on these relationships.
The experimentally determined PPIs were then used with AlphaFold-Multimer to construct high-quality structural models for 130 binary protein assemblies. The study identified PdhI as an inhibitor of pyruvate dehydrogenase and established a link between glycolysis and the Krebs cycle.
The researchers have emphasized the power of combining complementary techniques, such as in-cell crosslinking, to identify direct PPIs with high confidence without genetic alteration and to predict and verify associated structural models reliably.
Crosslinking Mass Spectroscopy
Using crosslinking mass spectrometry on B. subtilis cells, the scientists constructed a network of cell-wide interactions. They crosslinked proteins using a membrane-permeable crosslinker and discovered 560 protein interactions with less than a 2% false discovery rate for protein-protein interactions. There are 337 distinct proteins in the interaction network and 629 proteins with only self-links discovered.
Protein abundance was a crucial component in determining whether or not a protein could be identified with crosslinks, and some very abundant proteins provided several interactions to the method. The majority of crosslinks were within the predicted distance, as determined by the authors’ comparison of their data to known structures. After removing the interactions with the extremely prevalent proteins, 310 interactions, including 186 novel interactions, were found.
Two interactors, YugI and YabR that interact with several 30S ribosomal subunit proteins stood out among the many proteins discovered in the study with crosslinks to ribosomal subunits. Two-hybrid assays on bacteria validated their interactions with certain ribosomal proteins. In addition, the researchers identified 33 uncharacterized proteins in the crosslinking MS network that were overlooked in earlier research. Using size exclusion chromatography, the researchers compared protein complexes from untreated and crosslinked cells to see if the fixation of these interactions within the cell by crosslinking was crucial (SEC).
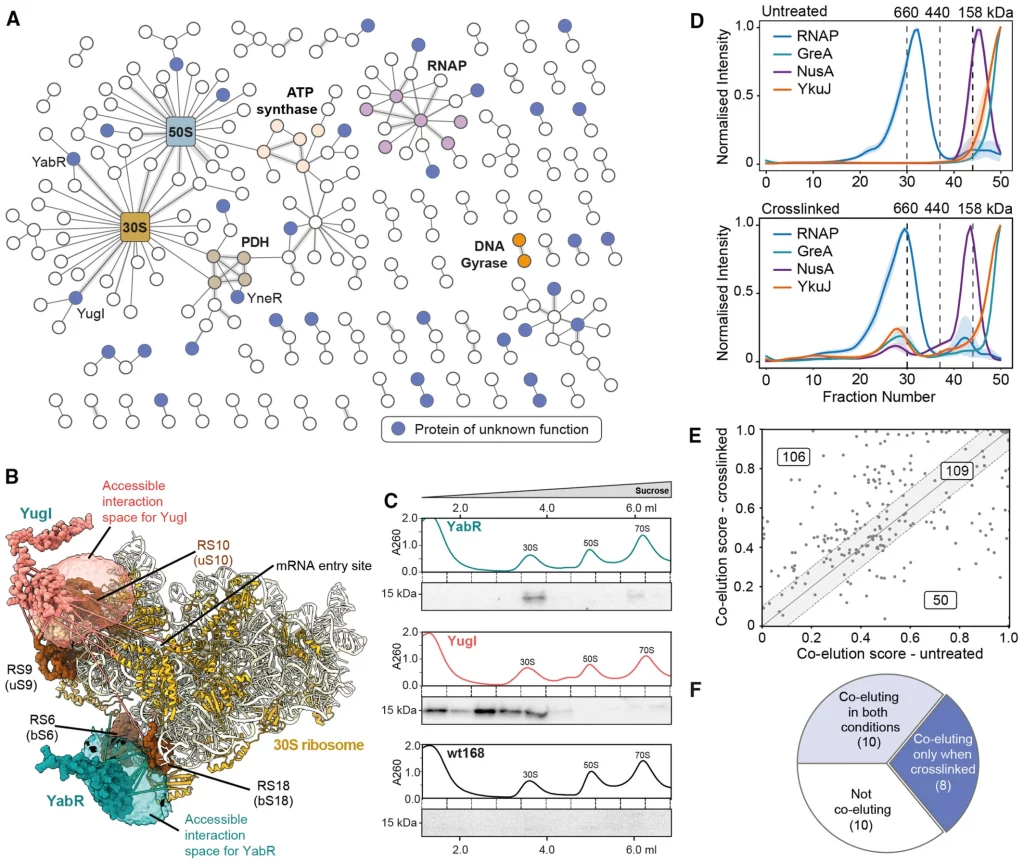
Image Source: https://doi.org/10.15252/msb.202311544
Size Exclusion Chromatography (SEC) and CoFrac-MS
In a second approach for detecting protein-protein interactions (PPIs), the researchers segregated the soluble proteomes of crosslinked and untreated B. subtilis cells using size exclusion chromatography (SEC). Crosslinking stabilized several components of known complexes and facilitated their co-elution, and a similar effect was reported for the uncharacterized protein YkuJ. More than two-thirds of the PPIs had a higher co-elution score following in-cell crosslinking, as determined by the researchers’ evaluation of the overall advantage of crosslinking in stabilizing protein complexes prior to cell lysis.
CoFrac-MS for finding protein-protein interactions (PPIs) requires proteome stabilization prior to cell lysis in order to discover interactions involving uncharacterized proteins. By comparing crosslinked and untreated circumstances, the scientists were able to increase the proportion of discovered PPIs from 36% to 62%. They then analyzed co-elution behavior using PCprophet and discovered 667 potential PPIs, 449 from cross-linked cells, and 318 from untreated cells. The CoFrac-MS candidate PPIs were supplementary to the crosslinking MS candidate PPIs, bringing the total number of PPIs and candidate PPIs to 878. The newly found PPIs have been uploaded to the SubtiWiki database.
Comprehensive modeling using AlphaFold-Multimer and SubtiWiki database
AlphaFold-Multimer was used to generate a comprehensive list of protein-protein interactions (PPIs) for system-wide structural modeling. Using high-confidence protein interactions from the manually maintained SubtiWiki database, the authors added known PPIs without structural information to their experimentally determined PPIs. They eliminated intra-ribosome interactions and homodimers with Protein Data Bank homologs, resulting in a final list of 1,218 previously identified PPIs and 2,032 prospective PPIs. The low overlap across the three datasets demonstrates the complementarity of the various methodologies.
Predicting high-confidence models using AlphaFold-Multimer
Authors utilized AlphaFold-Multimer to create structural models of previously established and newly discovered protein-protein interactions (PPIs). They evaluated the quality of the models using the total predicted TM score (pTM) and the predicted TM score at the interface (ipTM). They discovered 153 PPI models with high confidence (ipTM>0.85), including 17 novel interactions. Crosslinking MS had the greatest hit rate for the structural modeling of new PPIs among the three techniques (crosslinking MS, CoFrac-MS, and SubtiWiki).
The ipTM score indicates the quality of protein-protein interaction models produced by AlphaFold-Multimer. However, low ipTM values may not necessarily mean a lack of interaction between two proteins. It is important to bear these limitations in mind while reading AlphaFold-Multimer prediction results.
Heteromeric Crosslink Data
To validate AlphaFold models of protein complexes, the authors employed high-quality crosslink data. They discovered a substantial association between the ipTM score and the restraint satisfaction of heteromeric crosslinks, showing that high-confidence models match the observed residue-residue distances in situ. Crosslinking MS data can independently corroborate anticipated interfaces, particularly for PPIs with a high number of heteromeric crosslinks, where a large percentage of the interface is covered by crosslinking MS data.
Network Analysis
Due to its limits, network analysis can only give indirect information on interactions of higher order. Predicted binary interactions may be separate occurrences or components of larger multiprotein complexes. To discover probable higher-order complexes, PPI predictions with ipTM>0.65 were displayed as a network, leading to the identification of 64 groups containing between three and sixteen members. Functionally related proteins involved in DNA replication and recombination and sugar transport through the phosphotransferase system formed the biggest complexes.
To simplify the issue, probable heteromeric trimers with a 1:1:1 stoichiometry were predicted, yielding 33 groups of just three proteins, including five potential complexes with unique interactions. AlphaFold-Multimer was utilized to predict 14 trimer complexes, such as the lactate utilization operon LutA-LutB-LutC and the CapBCA complex involved in the production and export of c-polyglutamate. The membrane-spanning structure of the competence proteins ComEC-ComFC-ComFA suggests that the energy provided by ComFA-mediated ATP hydrolysis feeds ComEC-mediated absorption of single-stranded DNA molecules.
Conclusion
The paper discussed in this article presents genetic-free methodologies for screening protein-protein interactions in B. subtilis, which resulted in the identification of 114 high-quality binary interactions with no prior structural similarity. The technique is very effective for membrane proteins, and models with high scores exhibit a high level of crosslink distance restraint satisfaction. Therefore, models with low ipTM do not always indicate that they are not genuine interactors. Many novel biologically significant connections were discovered, such as YtpR in complex with the B subunit of glutamyl-tRNA amidotransferase (GatB), which shows a conserved and functionally significant association among prokaryotic species.
Article Source: Reference Paper
Learn More:




Sejal is a consulting scientific writing intern at CBIRT. She is an undergraduate student of the Department of Biotechnology at the Indian Institute of Technology, Kharagpur. She is an avid reader, and her logical and analytical skills are an asset to any research organization.





{kind=link}