

A collaborative effort between several medical departments at the University of Oslo led to the development of Cellsnake, a convenient tool for users to analyze single-cell RNA sequencing (scRNA-seq) data in an integrative manner, compatible with both Python and R. It can be incorporated into pre-existing workflow pipelines and can rapidly analyze several samples at once. It is open-source software, has a user-friendly interface for researchers, brings lower costs, and can be accessed through GitHub, PyPi, Docker, and Bioconda. scRNA-seq is a collection of transcriptome data at high resolution. By studying single cells, we can unlock intriguing heterogeneous characteristics present within populations of cells. While single cells can be analyzed using computational tools, they have many issues associated with them: they lack critical functions and modes for analyzing data in batches, and it can be difficult for users who lack computational expertise to install the required software.
An introduction to scRNA-seq
In simple words, in scRNA-seq, gene expression is studied at the level of just a single cell. Conventional RNA sequencing, which is carried out in bulk, studies the average of the overall transcription expression within cell populations. scRNA was initially used in a limited capacity due to high costs and technical complexities. With advancements in computational technology over the past few decades, scientists have found greater accessibility to performing and studying high-level scRNA-seq analysis. Such access will provide crucial information related to the various states exhibited by cells, the interactions between cells, and the identification of novel cells.
Any standard workflow related to scRNA-seq data involves the following steps: filtering of data, normalization, scaling, reduction of dimensionality, clustering, visualization, analysis of differential expressions, analysis of functions, and finally, annotation of data. Some of the popularly used workflows for scRNA-seq include Seurat and Scanpy.
Challenges in scRNA-seq analysis
Scientists have faced many technical issues when analyzing scRNA-seq data, such as the presence of dead cells, low counts for unique molecular identifiers (UMI), unwanted side effects of batch analysis, low levels of expression, and high-dimensional structures of data. All of these have been barriers to advancing single-cell research. To tackle all these shortcomings, there have been attempts to develop novel bioinformatics tools that can be integrated into existing workflow pipelines. A major problem with many of these existing bioinformatics tools, however, is that they require prior knowledge related to computation, which results in limiting the number of researchers that can utilize them and, therefore, slows down progress in this domain.
Another issue comes with multiple RNA-seq datasets. While the process of analyzing samples is thoroughly documented and recorded with all parameters intact for researchers to refer to, it can still be challenging to keep track of each and every step taken throughout the process. This becomes increasingly difficult when more than one sample is available. Utilizing the power of high-performance computing (HPC) can be a hassle as well. Existing workflows for batch and differential expression analysis are either highly specialized or have limited functions available for use.
Introducing Cellsnake
Cellsnake is a pipeline-type workflow that is independent of platforms and acts as a command-line application. It provides accessibility, flexibility, and reproducibility to users. An important feature to note is its ability to make use of various scRNA-seq algorithms to make tasks like automatic mitochondrial (MT) gene trimming, optimal clustering selection at a high resolution, filtering of doublets, visualization of marker genes, and analysis of pathways much simpler and more convenient than they were before. It is compatible with HPC platforms as well.
Some additional features of Cellsnake include the provision of metagenome analysis in cases where unmapped reads are present and the generation of intermediate files (for example, data files from R), which can be used for extraction, storage, reproducibility, and sharing purposes. It also contains automated tools for performing automated downstream analysis. Pre-processing functions include essential steps such as quality control (QC), filtering, and automatic selection of parameters. Cellsnake also helps in identifying pathways for specific genes within cell subpopulations. It can also integrate multiple scRNA-seq datasets for the identification of cell types that are shared or unique. From a reproducibility point of view, Cellsnake creates separate folders to restrict different versions of tools within a certain environment and only allow access to specific tools when required. Configuration files are saved in their own version within the workflow; storage of downstream analysis data is set to default; and for each version in the Docker repository, different images are distributed within the workflow.
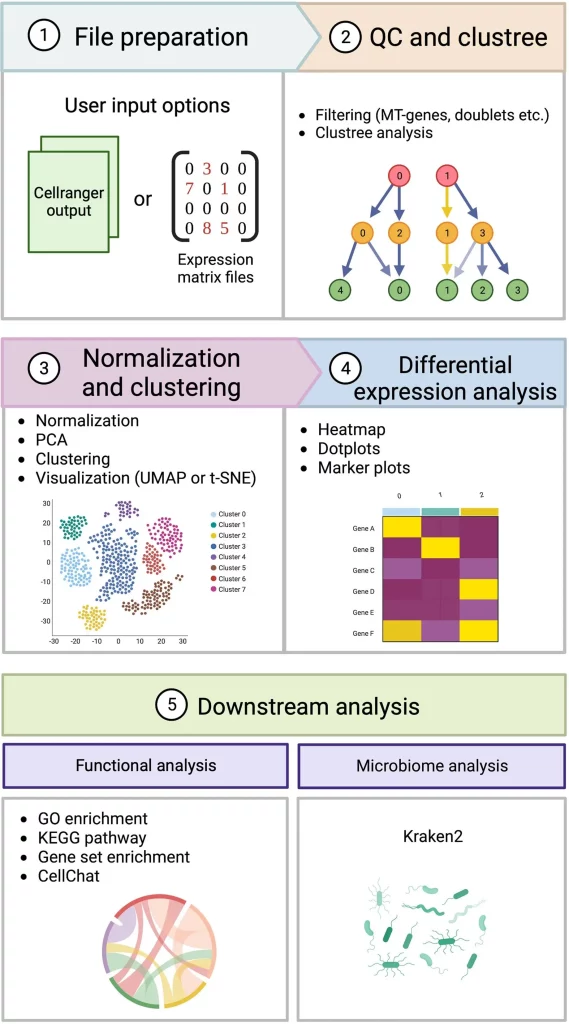
Image Source: https://doi.org/10.1093/gigascience/giad091
Advantages offered by Cellsnake
A vast range of tools are available for use within Cellsnake. It can run on local computers as well as HPCs, with enhanced data sharing and accessibility made possible through the serialization of R data. Cellsnake has made discoveries related to the codependent relationship between microbial heterogeneity and the types of cells microbes are associated with. The reads for these characteristics can be found within the scRNA-seq data. Cellsnake uses Kraken2 to analyze this information, and parameters are fine-tuned to increase specificity and cater to the personal databases of the user. It should be noted that microbes may not always originate within tissues and cells, and they may come from external contamination sources as well. Nevertheless, it can still be a useful guide to help scientists identify microbial sources that contaminate samples.
Limitations of Cellsnake
Cellsnake requires a large portion of disc space for storing metadata files that are used for performing advanced downstream analysis in later stages. It heavily relies on outputs given by the 10X Genomics platform, but this information may not always be available for the user at all times. It also has a slow rate of auto-detecting key parameters when a large number of cells are involved. To improve this aspect, a parallel version of the MultiK tool was used by the researchers.
Conclusion
Cellsnake makes scRNA-seq data analysis customizable and reproducible for researchers in a highly convenient and user-friendly manner. It follows a streamlined workflow pattern and can transform research domains within single-cell-level gene expression studies as we know them.
Article source: Reference Paper | Cellsnake Availability: PyPi | Bioconda | Docker | Snakemake workflow | Documentation
Learn More:




Swasti is a scientific writing intern at CBIRT with a passion for research and development. She is pursuing BTech in Biotechnology from Vellore Institute of Technology, Vellore. Her interests deeply lie in exploring the rapidly growing and integrated sectors of bioinformatics, cancer informatics, and computational biology, with a special emphasis on cancer biology and immunological studies. She aims to introduce and invest the readers of her articles to the exciting developments bioinformatics has to offer in biological research today.











{kind=link}